
Primary Human Blood Dendritic Cells for Cancer Immunotherapy—Tailoring the Immune Response by Dendritic Cell Maturation


Abstract
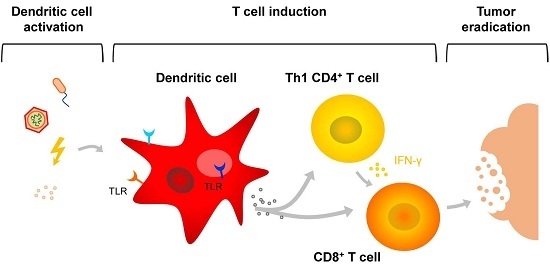
:
{kind=link}
{kind=link}
1. Introduction
2. Dendritic Cell Maturation
3. Monocyte Derived DC Maturation for Cancer Immunotherapy
4. Maturation of Plasmacytoid DCs in the Context of Cancer Immunotherapy
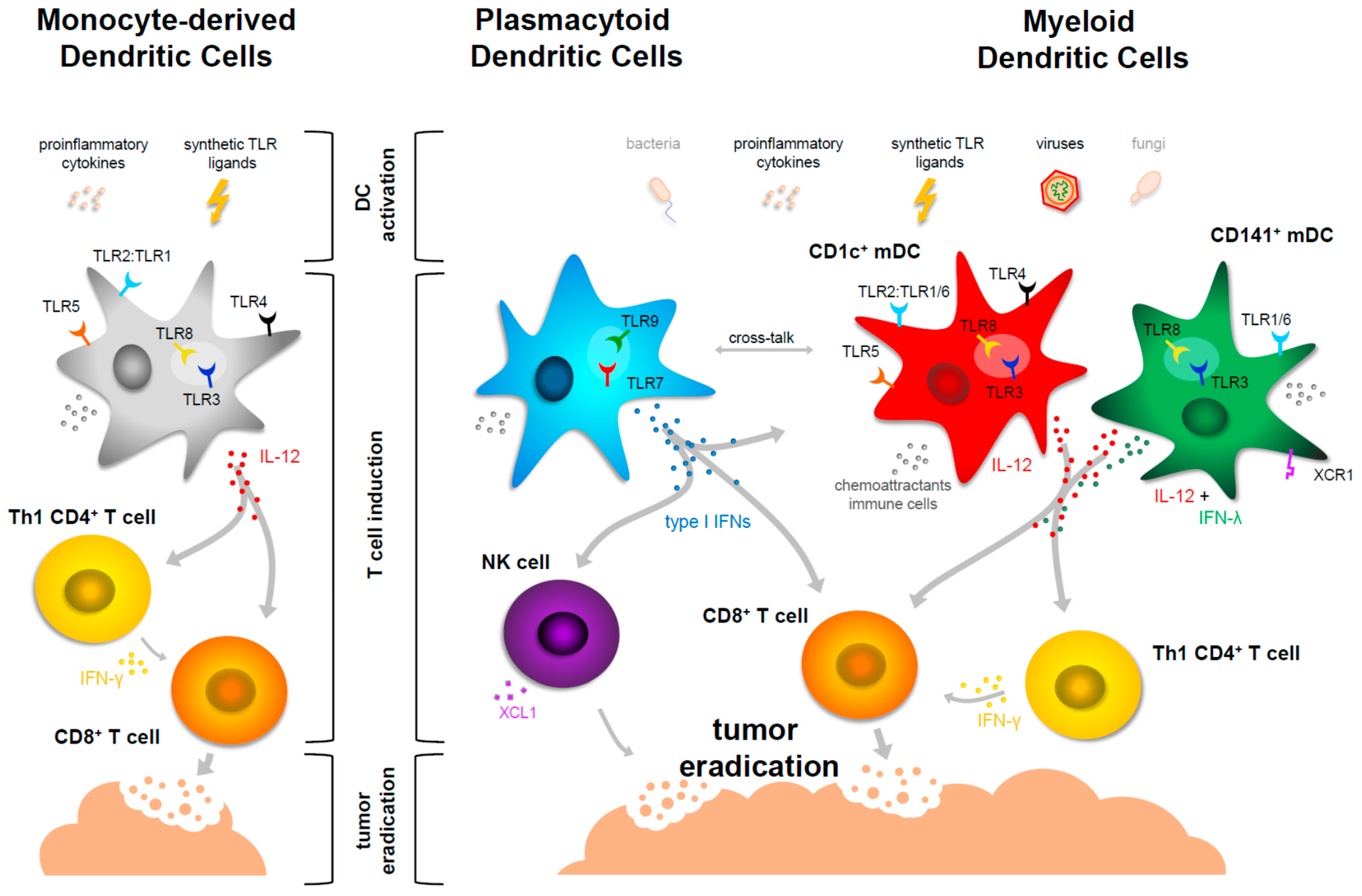
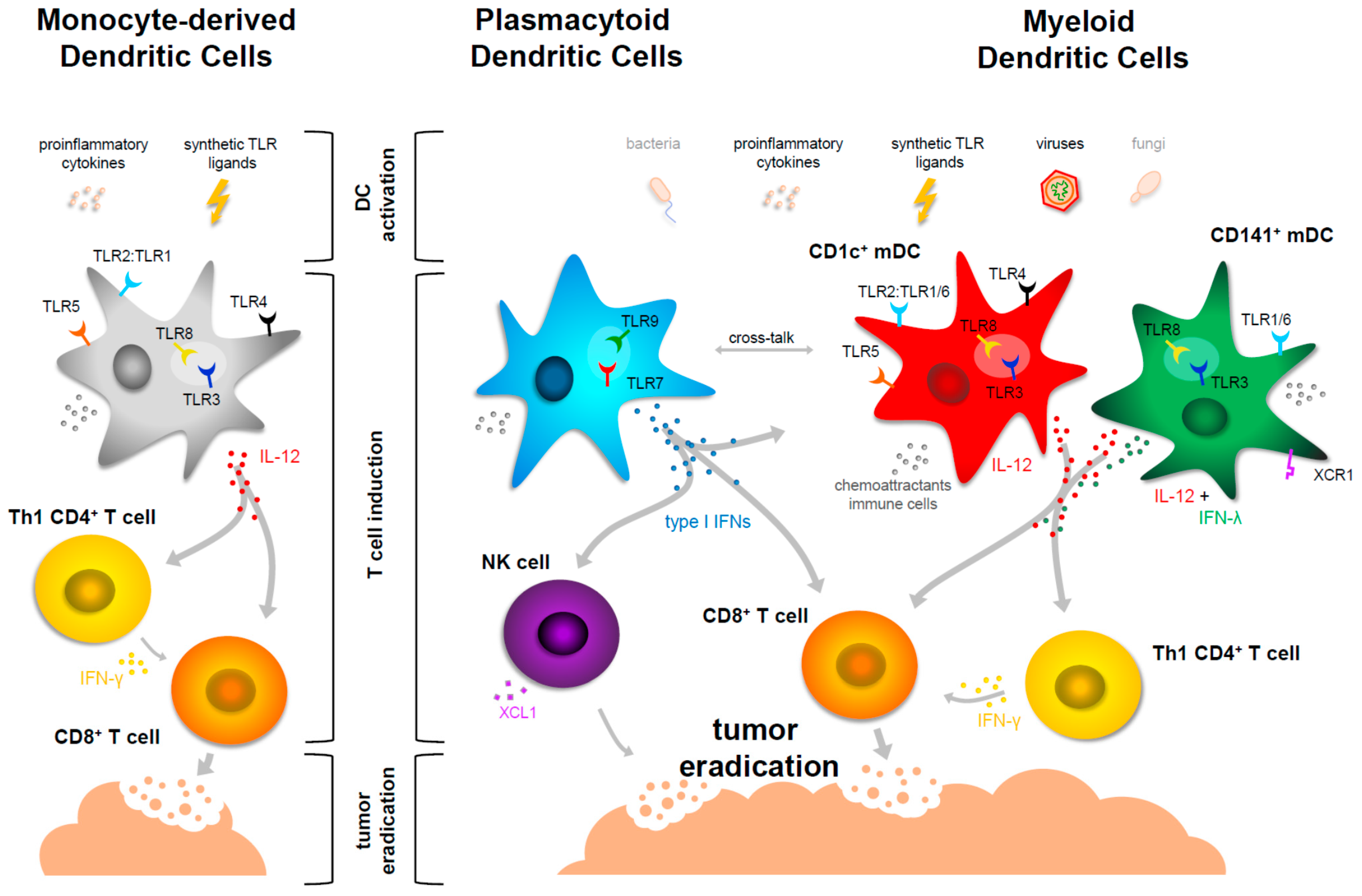
5. Maturation of Myeloid DCs in the Context of Cancer Immunotherapy
6. Human Primary DC Subsets as Cancer Vaccines
7. Inducing Maturation of DC Subsets by in Vivo Targeting
8. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Den Brok, M.H.M.G.M.; Nierkens, S.; Figdor, C.G.; Ruers, T.J.M.; Adema, G.J. Dendritic cells: Tools and targets for antitumor vaccination. Expert Rev. Vaccines 2005, 4, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Tuyaerts, S.; Aerts, J.L.; Corthals, J.; Neyns, B.; Heirman, C.; Breckpot, K.; Thielemans, K.; Bonehill, A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol. Immunother. 2007, 56, 1513–1537. [Google Scholar] [CrossRef] [PubMed]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three markers for distinct subsets of dendritic cells in human peripheral blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Sallusto, F. Regulation of T cell immunity by dendritic cells. Cell 2001, 106, 263–266. [Google Scholar] [CrossRef]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M. Intestinal CD103+ dendritic cells: Master regulators of tolerance? Trends Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-Like receptors (TLRs) and their ligands. Handb. Exp. Pharmacol. 2008, 183, 1–20. [Google Scholar] [PubMed]
- Bakdash, G.; Sittig, S.P.; van Dijk, T.; Figdor, C.G.; de Vries, I.J.M. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and its role in the generation of TH1 cells. Immunol. Today 1993, 14, 335–338. [Google Scholar] [CrossRef]
- Levings, M.K.; Sangregorio, R.; Galbiati, F.; Squadrone, S.; de Waal Malefyt, R.; Roncarolo, M.-G. IFN-α and IL-10 Induce the Differentiation of Human Type 1 T Regulatory Cells. J. Immunol. 2001, 166, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Reider, D.; Heuer, M.; Ebner, S.; Kämpgen, E.; Eibl, B.; Niederwieser, D.; Schuler, G. Generation of mature dendritic cells from human blood An improved method with special regard to clinical applicability. J. Immunol. Methods 1996, 196, 137–151. [Google Scholar] [CrossRef]
- Bender, A.; Sapp, M.; Schuler, G.; Steinman, R.M.; Bhardwaj, N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods 1996, 196, 121–135. [Google Scholar] [CrossRef]
- Jonuleit, H.; Giesecke, A.; Kandemir, A.; Paragnik, L.; Knop, J.; Enk, A.H. Induction of tumor peptide-specific cytotoxic T cells under serum-free conditions by mature human dendritic cells. Arch. Dermatol. Res. 2000, 292, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Giesecke-Tuettenberg, A.; Tüting, T.; Thurner-Schuler, B.; Stuge, T.B.; Paragnik, L.; Kandemir, A.; Lee, P.P.; Schuler, G.; Knop, J.; et al. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int. J. Cancer 2001, 93, 243–251. [Google Scholar] [CrossRef] [PubMed]
- McIlroy, D.; Gregoire, M. Optimizing dendritic cell-based anticancer immunotherapy: Maturation state does have clinical impact. Cancer Immunol. Immunother. 2003, 52, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Draube, A.; Klein-González, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Iyoda, T.; Tarbell, K.; Olson, K.; Velinzon, K.; Inaba, K.; Steinman, R.M. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 2003, 198, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Loschko, J.; Heink, S.; Hackl, D.; Dudziak, D.; Reindl, W.; Korn, T.; Krug, A.B. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J. Immunol. 2011, 187, 6346–6356. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Kaliński, P.; Schuitemaker, J.H.; Hilkens, C.M.; Kapsenberg, M.L. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: The levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 1998, 161, 2804–2809. [Google Scholar] [PubMed]
- Harizi, H.; Juzan, M.; Pitard, V.; Moreau, J.-F.; Gualde, N. Cyclooxygenase-2-issued prostaglandin E2 enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 2002, 168, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Muthuswamy, R.; Mueller-Berghaus, J.; Haberkorn, U.; Reinhart, T.A.; Schadendorf, D.; Kalinski, P. PGE2 transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 2010, 116, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Kaliński, P.; Vieira, P.L.; Schuitemaker, J.H.; de Jong, E.C.; Kapsenberg, M.L. Prostaglandin E2 is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood 2001, 97, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Muthuswamy, R.; Urban, J.; Lee, J.-J.; Reinhart, T.A.; Bartlett, D.; Kalinski, P. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 2008, 68, 5972–5978. [Google Scholar] [CrossRef] [PubMed]
- Boullart, A.C.I.; Aarntzen, E.H.J.G.; Verdijk, P.; Jacobs, J.F.M.; Schuurhuis, D.H.; Benitez-Ribas, D.; Schreibelt, G.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.; et al. Maturation of monocyte-derived dendritic cells with Toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol. Immunother. 2008, 57, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Javorovic, M.; Bürdek, M.; Wilde, S.; Mosetter, B.; Tippmer, S.; Bigalke, I.; Geiger, C.; Schendel, D.J.; Frankenberger, B. Generation of Th1-polarizing dendritic cells using the TLR7/8 agonist CL075. J. Immunol. 2010, 185, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Dauer, M.; Lam, V.; Arnold, H.; Junkmann, J.; Kiefl, R.; Bauer, C.; Schnurr, M.; Endres, S.; Eigler, A. Combined use of toll-like receptor agonists and prostaglandin E2 in the FastDC model: Rapid generation of human monocyte-derived dendritic cells capable of migration and IL-12p70 production. J. Immunol. Methods 2008, 337, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Korthals, M.; Safaian, N.; Kronenwett, R.; Maihöfer, D.; Schott, M.; Papewalis, C.; Diaz Blanco, E.; Winter, M.; Czibere, A.; Haas, R.; et al. Monocyte derived dendritic cells generated by IFN-alpha acquire mature dendritic and natural killer cell properties as shown by gene expression analysis. J. Transl. Med. 2007, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Severa, M.; Remoli, M.E.; Giacomini, E.; Ragimbeau, J.; Lande, R.; Uzé, G.; Pellegrini, S.; Coccia, E.M. Differential responsiveness to IFN-α and IFN-β of human mature DC through modulation of IFNAR expression. J. Leukoc. Biol. 2006, 79, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.M.; Lapenta, C.; Santodonato, L.; D’Agostino, G.; Belardelli, F.; Ferrantini, M. Dendritic Cells; Lombardi, G., Riffo-Vasquez, Y., Eds.; Handbook of Experimental Pharmacology; Springer: Heidelberg, Germany, 2009; Volume 188. [Google Scholar]
- Pan, J.; Zhang, M.; Wang, J.; Wang, Q.; Xia, D.; Sun, W.; Zhang, L.; Yu, H.; Liu, Y.; Cao, X. Interferon-gamma is an autocrine mediator for dendritic cell maturation. Immunol. Lett. 2004, 94, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wesa, A.; Kalinski, P.; Kirkwood, J.M.; Tatsumi, T.; Storkus, W.J. Polarized Type-1 Dendritic Cells (DC1) Producing High Levels of IL-12 Family Members Rescue Patient TH1-type Antimelanoma CD4+ T cell Responses In Vitro. J. Immunother. 2007, 30, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [PubMed]
- Jarrossay, D.; Napolitani, G.; Colonna, M.; Sallusto, F.; Lanzavecchia, A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 3388–3393. [Google Scholar] [CrossRef]
- Kadowaki, N.; Ho, S.; Antonenko, S.; de Waal Malefyt, R.; Kastelein, R.A.; Bazan, F.; Liu, Y.-J. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. J. Exp. Med. 2001, 194, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 mRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Tel, J.; Sliepen, K.H.E.W.J.; Benitez-Ribas, D.; Figdor, C.G.; Adema, G.J.; de Vries, I.J.M. Toll-like receptor expression and function in human dendritic cell subsets: Implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, A.; Asselin-Paturel, C.; Gilliet, M.; Crain, C.; Trinchieri, G.; Liu, Y.-J.; O’Garra, A. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: Dependency on antigen dose and differential toll-like receptor ligation. J. Exp. Med. 2003, 197, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Mathan, T.S.M.; Figdor, C.G.; Buschow, S.I. Human plasmacytoid dendritic cells: From molecules to intercellular communication network. Front. Immunol. 2013, 4, 372. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010, 234, 142–162. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Schreibelt, G.; Sittig, S.P.; Mathan, T.S.M.; Buschow, S.I.; Cruz, L.J.; Lambeck, A.J.A.; Figdor, C.G.; de Vries, I.J.M. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013, 121, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; van der Leun, A.M.; Figdor, C.G.; Torensma, R.; de Vries, I.J.M. Harnessing human plasmacytoid dendritic cells as professional APCs. Cancer Immunol. Immunother. 2012, 61, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ripoche, A.-C.; Matheoud, D.; Nascimbeni, M.; Escriou, N.; Lebon, P.; Heshmati, F.; Guillet, J.-G.; Gannagé, M.; Caillat-Zucman, S.; et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity 2007, 27, 481–92. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sörensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.; Manches, O.; Angel, J.; Molens, J.-P.; Chaperot, L.; Plumas, J. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS ONE 2009, 4, e7111. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Chen, Q.; Shin, A.; Chen, W.; Toy, T.; Jenderek, C.; Green, S.; Miloradovic, L.; Drane, D.; Davis, I.D.; et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood 2005, 105, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Mittag, D.; Proietto, A.I.; Loudovaris, T.; Mannering, S.I.; Vremec, D.; Shortman, K.; Wu, L.; Harrison, L.C. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J. Immunol. 2011, 186, 6207–6217. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Young, L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 2008, 29, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Lambeck, A.J.A.; Cruz, L.J.; Tacken, P.J.; de Vries, I.J.M.; Figdor, C.G. Human plasmacytoid dendritic cells phagocytose, process, and present exogenous particulate antigen. J. Immunol. 2010, 184, 4276–4283. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi, I.; Morandi, B.; Antsiferova, O.; Costa, G.; Oliveri, D.; Conte, R.; Pezzino, G.; Vermiglio, G.; Anastasi, G.P.; Navarra, G.; et al. Membrane transfer from tumor cells overcomes deficient phagocytic ability of plasmacytoid dendritic cells for the acquisition and presentation of tumor antigens. J. Immunol. 2014, 192, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Montoya, M.; Schiavoni, G.; Mattei, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P. Human natural interferon-alpha producing cells. Pharmacol. Ther. 1993, 60, 39–62. [Google Scholar] [CrossRef]
- Montoya, C.J.; Jie, H.-B.; Al-Harthi, L.; Mulder, C.; Patino, P.J.; Rugeles, M.T.; Krieg, A.M.; Landay, A.L.; Wilson, S.B. Activation of Plasmacytoid Dendritic Cells with TLR9 Agonists Initiates Invariant NKT Cell-Mediated Cross-Talk with Myeloid Dendritic Cells. J. Immunol. 2006, 177, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, X.; Tough, D.F.; Sprent, J. Type I interferon-mediated stimulation of T cells by CpG DNA. J. Exp. Med. 1998, 188, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Kappler, J.; Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999, 189, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, Y.; Lizée, G.; Qin, H.; Liu, S.; Rabinovich, B.; Kim, G.J.; Wang, Y.-H.; Ye, Y.; Sikora, A.G.; et al. Plasmacytoid dendritic cells induce NK cell—Dependent, tumor antigen—Specific T cell cross-priming and tumor regression in mice. J. Clin. Invest. 2008, 118, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Bacher, N.; Graulich, E.; Jonuleit, H.; Grabbe, S.; Steinbrink, K. Interferon-α abrogates tolerance induction by human tolerogenic dendritic cells. PLoS ONE 2011, 6, e22763. [Google Scholar] [CrossRef] [PubMed]
- Nierkens, S.; den Brok, M.H.; Garcia, Z.; Togher, S.; Wagenaars, J.; Wassink, M.; Boon, L.; Ruers, T.J.; Figdor, C.G.; Schoenberger, S.P.; et al. Immune adjuvant efficacy of CpG oligonucleotide in cancer treatment is founded specifically upon TLR9 function in plasmacytoid dendritic cells. Cancer Res. 2011, 71, 6428–6437. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Machelon, V.; Coulomb-L’Hermin, A.; Borvak, J.; Nome, F.; Isaeva, T.; Wei, S.; Krzysiek, R.; Durand-Gasselin, I.; Gordon, A.; et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 2001, 7, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Treilleux, I.; Blay, J.-Y.; Bendriss-Vermare, N.; Ray-Coquard, I.; Bachelot, T.; Guastalla, J.-P.; Bremond, A.; Goddard, S.; Pin, J.-J.; Barthelemy-Dubois, C.; et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res. 2004, 10, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, E.; Wollenberg, B.; Rothenfusser, S.; Wagner, M.; Wellisch, D.; Mack, B.; Giese, T.; Gires, O.; Endres, S.; Hartmann, G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003, 63, 6478–6487. [Google Scholar] [PubMed]
- Salio, M.; Cella, M.; Vermi, W.; Facchetti, F.; Palmowski, M.J.; Smith, C.L.; Shepherd, D.; Colonna, M.; Cerundolo, V. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur. J. Immunol. 2003, 33, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Menetrier-Caux, C.; Montmain, G.; Dieu, M.C.; Bain, C.; Favrot, M.C.; Caux, C.; Blay, J.Y. Inhibition of the differentiation of dendritic cells from CD34+ progenitors by tumor cells: Role of interleukin-6 and macrophage colony-stimulating factor. Blood 1998, 92, 4778–4791. [Google Scholar] [PubMed]
- Bell, D.; Chomarat, P.; Broyles, D.; Netto, G.; Harb, G.M.; Lebecque, S.; Valladeau, J.; Davoust, J.; Palucka, K.A.; Banchereau, J. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J. Exp. Med. 1999, 190, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Kors, C.; Audring, H.; Walden, P.; Sterry, W.; Trefzer, U. Phase 1 Evaluation of Intralesionally Injected TLR9-agonist PF-3512676 in Patients With Basal Cell Carcinoma or Metastatic Melanoma. J. Immunother. 2008, 31, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Urosevic, M.; Kempf, W.; Hoek, K.; Hafner, J.; Burg, G. Imiquimod in basal cell carcinoma: How does it work? Br. J. Dermatol. 2003, 149, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Heer, H.J.; Hammad, H.; Soullié, T.; Hijdra, D.; Vos, N.; Willart, M.A.M.; Hoogsteden, H.C.; Lambrecht, B.N. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 2004, 200, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Hemont, C.; Neel, A.; Heslan, M.; Braudeau, C.; Josien, R. Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J. Leukoc. Biol. 2013, 93, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Nizzoli, G.; Krietsch, J.; Weick, A.; Steinfelder, S.; Facciotti, F.; Gruarin, P.; Bianco, A.; Steckel, B.; Moro, M.; Crosti, M.; et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 2013, 122, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; Vulink, A.J.E.; Hart, D.N.J.; Radford, K.J. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Chatterjee, B.; Esselborn, F.; Smed-Sorensen, A.; Nakamura, N.; Chalouni, C.; Lee, B.-C.; Vandlen, R.; Keler, T.; Lauer, P.; et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 2013, 210, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Piccioli, D.; Tavarini, S.; Borgogni, E.; Steri, V.; Nuti, S.; Sammicheli, C.; Bardelli, M.; Montagna, D.; Locatelli, F.; Wack, A. Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood 2007, 109, 5371–5379. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, M.; Lundberg, K.; Borrebaeck, C.A.K. Gene family clustering identifies functionally associated subsets of human in vivo blood and tonsillar dendritic cells. J. Immunol. 2005, 175, 4839–4846. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, H.; Bathke, B.; Gilles, S.; Traidl-Hoffmann, C.; Luber, C.A.; Fejer, G.; Freudenberg, M.A.; Davey, G.M.; Vremec, D.; Kallies, A.; et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J. Exp. Med. 2010, 207, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.-N.; Malinarich, F.; Malleret, B.; et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Bacon, K.B.; Premack, B.A.; Gardner, P.; Schall, T.J. Activation of dual T cell signaling pathways by the chemokine RANTES. Science 1995, 269, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Meixlsperger, S.; Leung, C.S.; Ramer, P.C.; Pack, M.; Vanoaica, L.D.; Breton, G.; Pascolo, S.; Salazar, A.M.; Dzionek, A.; Schmitz, J.; et al. CD141+ dendritic cells produce prominent amounts of IFN-α after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood 2013, 121, 5034–5044. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Edwards, A.D.; Schito, M.; Aliberti, J.; Manickasingham, S.; Sher, A.; Reis e Sousa, C. CD40 Triggering of Heterodimeric IL-12 p70 Production by Dendritic Cells in vivo Requires a Microbial Priming Signal. Immunity 2000, 13, 453–462. [Google Scholar] [CrossRef]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Sittig, S.P.; Bakdash, G.; Weiden, J.; Sköld, A.E.; Tel, J.; Figdor, C.G.; de Vries, I.J.M.; Schreibelt, G. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediat. Inflamm. 2015. under revision. [Google Scholar]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Perrot, I.; Deauvieau, F.; Massacrier, C.; Hughes, N.; Garrone, P.; Durand, I.; Demaria, O.; Viaud, N.; Gauthier, L.; Blery, M.; et al. TLR3 and Rig-like receptor on myeloid dendritic cells and Rig-like receptor on human NK cells are both mandatory for production of IFN-gamma in response to double-stranded RNA. J. Immunol. 2010, 185, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Guiton, R.; Contreras, V.; Feuillet, V.; Dutertre, C.-A.; Ventre, E.; Vu Manh, T.-P.; Baranek, T.; Storset, A.K.; Marvel, J.; et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Valladeau-Guilemond, J.; Donnadieu, M.-H.M.-H.; Sastre-Garau, X.; Soumelis, V.; Amigorena, S. Characterization of resident and migratory dendritic cells in human lymph nodes. J. Exp. Med. 2012, 209, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Kassianos, A.J.; Hardy, M.Y.; Ju, X.; Vijayan, D.; Ding, Y.; Vulink, A.J.E.; McDonald, K.J.; Jongbloed, S.L.; Wadley, R.B.; Wells, C.; et al. Human CD1c (BDCA-1)+ myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to Escherichia coli. Eur. J. Immunol. 2012, 42, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Huysamen, C.; Willment, J.A.; Dennehy, K.M.; Brown, G.D. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 2008, 283, 16693–16701. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; Reis e Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-López, R.; de Smedt, T.; Michel, P.; Godfroid, J.; Pajak, B.; Heirman, C.; Thielemans, K.; Leo, O.; Urbain, J.; Moser, M. CD8α+ and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 1999, 189, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Soruri, A.; Kiafard, Z.; Dettmer, C.; Riggert, J.; Köhl, J.; Zwirner, J. IL-4 down-regulates anaphylatoxin receptors in monocytes and dendritic cells and impairs anaphylatoxin-induced migration in vivo. J. Immunol. 2003, 170, 3306–3314. [Google Scholar] [CrossRef] [PubMed]
- Breckpot, K.; Corthals, J.; Bonehill, A.; Michiels, A.; Tuyaerts, S.; Aerts, C.; Heirman, C.; Thielemans, K. Dendritic cells differentiated in the presence of IFN-β and IL-3 are potent inducers of an antigen-specific CD8+ T cell response. J. Leukoc. Biol. 2005, 78, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.J.G.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.G.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.M.; Tel, J.; Benitez-Ribas, D.; Torensma, R.; Figdor, C.G. Prophylactic vaccines mimic synthetic CpG oligonucleotides in their ability to modulate immune responses. Mol. Immunol. 2011, 48, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Smits, E.L.; Anguille, S.; Joshi, R.N.; Figdor, C.G.; de Vries, I.J.M. Human plasmacytoid dendritic cells are equipped with antigen-presenting and tumoricidal capacities. Blood 2012, 120, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. 2015. conditionally accepted. [Google Scholar]
- Schreibelt, G.; Benitez-Ribas, D.; Schuurhuis, D.; Lambeck, A.J.A.; van Hout-Kuijer, M.; Schaft, N.; Punt, C.J.A.; Figdor, C.G.; Adema, G.J.; de Vries, I.J.M. Commonly used prophylactic vaccines as an alternative for synthetically produced TLR ligands to mature monocyte-derived dendritic cells. Blood 2010, 116, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Sköld, A.E.; van Beek, J.J.P.; Sittig, S.P.; Bakdash, G.; Tel, J.; Schreibelt, G.; de Vries, I.J.M. Protamine-stabilized RNA as an ex vivo stimulant of primary human dendritic cell subsets. Cancer Immunol. Immunother. 2015, 64, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Harty, J.T.; Badovinac, V.P. Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol. 2008, 8, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Schreurs, I.; Schreibelt, G.; Tel, J. Crosstalk between dendritic cell subsets and implications for dendritic cell-based anticancer immunotherapy. Expert Rev. Clin. Immunol. 2014, 10, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Vremec, D.; Ahmet, F.; Lahoud, M.H.; Villadangos, J.A.; Murphy, K.M.; Heath, W.R.; Shortman, K. Antibody responses initiated by Clec9A-bearing dendritic cells in normal and Batf3−/− mice. Mol. Immunol. 2012, 50, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Jahrsdörfer, B.; Weiner, G.J. CpG oligodeoxynucleotides as immunotherapy in cancer. Updat. Cancer Ther. 2008, 3, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Nierkens, S.; den Brok, M.H.; Roelofsen, T.; Wagenaars, J.A.L.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Route of administration of the TLR9 agonist CpG critically determines the efficacy of cancer immunotherapy in mice. PLoS ONE 2009, 4, e8368. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Zeelenberg, I.S.; Cruz, L.J.; van Hout-Kuijer, M.A.; van de Glind, G.; Fokkink, R.G.; Lambeck, A.J.A.; Figdor, C.G. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011, 118, 6836–6844. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front. Immunol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Sittig, S.P.; Blom, R.A.M.; Cruz, L.J.; Schreibelt, G.; Figdor, C.G.; de Vries, I.J.M. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J. Immunol. 2013, 191, 5005–5012. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Smed-Sörensen, A.; Cohn, L.; Chalouni, C.; Vandlen, R.; Lee, B.-C.; Widger, J.; Keler, T.; Delamarre, L.; Mellman, I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012, 120, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Kautz, A.; Böhnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Ribas, D.; Adema, G.J.; Winkels, G.; Klasen, I.S.; Punt, C.J.A.; Figdor, C.G.; de Vries, I.J.M. Plasmacytoid dendritic cells of melanoma patients present exogenous proteins to CD4+ T cells after Fc gamma RII-mediated uptake. J. Exp. Med. 2006, 203, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Flinsenberg, T.W.H.; Compeer, E.B.; Koning, D.; Klein, M.; Amelung, F.J.; van Baarle, D.; Boelens, J.J.; Boes, M. Fcγ receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood 2012, 120, 5163–5172. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.-I.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Fossum, E.; Grødeland, G.; Terhorst, D.; Tveita, A.A.; Vikse, E.; Mjaaland, S.; Henri, S.; Malissen, B.; Bogen, B. Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur. J. Immunol. 2014, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Sousa, C.R. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.Y.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.H.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–73. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Klinkenberg, L.J.J.; Cruz, L.J.; Tacken, P.J.; Tel, J.; Kreutz, M.; Adema, G.J.; Brown, G.D.; Figdor, C.G.; de Vries, I.J.M. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012, 119, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Sancho, D.; Zelenay, S.; Keller, A.M.; Reis e Sousa, C. Efficient and versatile manipulation of the peripheral CD4+ T-cell compartment by antigen targeting to DNGR-1/CLEC9A. Eur. J. Immunol. 2010, 40, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Khan, N.; Nakaya, H.I.; Li, S.; Loebbermann, J.; Maddur, M.S.; Park, Y.; Jones, D.P.; Chappert, P.; Davoust, J.; et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation—Supplementary material. Science 2014, 343, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Pierret, L.; Wilgenhof, S.; Corthals, J.; Roelandt, T.; Thielemans, K.; Neyns, B. Correlation between prior therapeutic dendritic cell vaccination and the outcome of patients with metastatic melanoma treated with ipilimumab. In 2009 ASCO Annual Meeting Proceedings (Post-Meeting Edition), Orlando, FL, USA, 29 May–2 June 2009; p. e20006.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sittig, S.P.; De Vries, I.J.M.; Schreibelt, G. Primary Human Blood Dendritic Cells for Cancer Immunotherapy—Tailoring the Immune Response by Dendritic Cell Maturation. Biomedicines 2015, 3, 282-303. https://doi.org/10.3390/biomedicines3040282
Sittig SP, De Vries IJM, Schreibelt G. Primary Human Blood Dendritic Cells for Cancer Immunotherapy—Tailoring the Immune Response by Dendritic Cell Maturation. Biomedicines. 2015; 3(4):282-303. https://doi.org/10.3390/biomedicines3040282
Chicago/Turabian StyleSittig, Simone P., I. Jolanda M. De Vries, and Gerty Schreibelt. 2015. "Primary Human Blood Dendritic Cells for Cancer Immunotherapy—Tailoring the Immune Response by Dendritic Cell Maturation" Biomedicines 3, no. 4: 282-303. https://doi.org/10.3390/biomedicines3040282
APA StyleSittig, S. P., De Vries, I. J. M., & Schreibelt, G. (2015). Primary Human Blood Dendritic Cells for Cancer Immunotherapy—Tailoring the Immune Response by Dendritic Cell Maturation. Biomedicines, 3(4), 282-303. https://doi.org/10.3390/biomedicines3040282