
Genetic Variants in Metabolic Pathways and Their Role in Cardiometabolic Risk: An Observational Study of >4000 Individuals

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
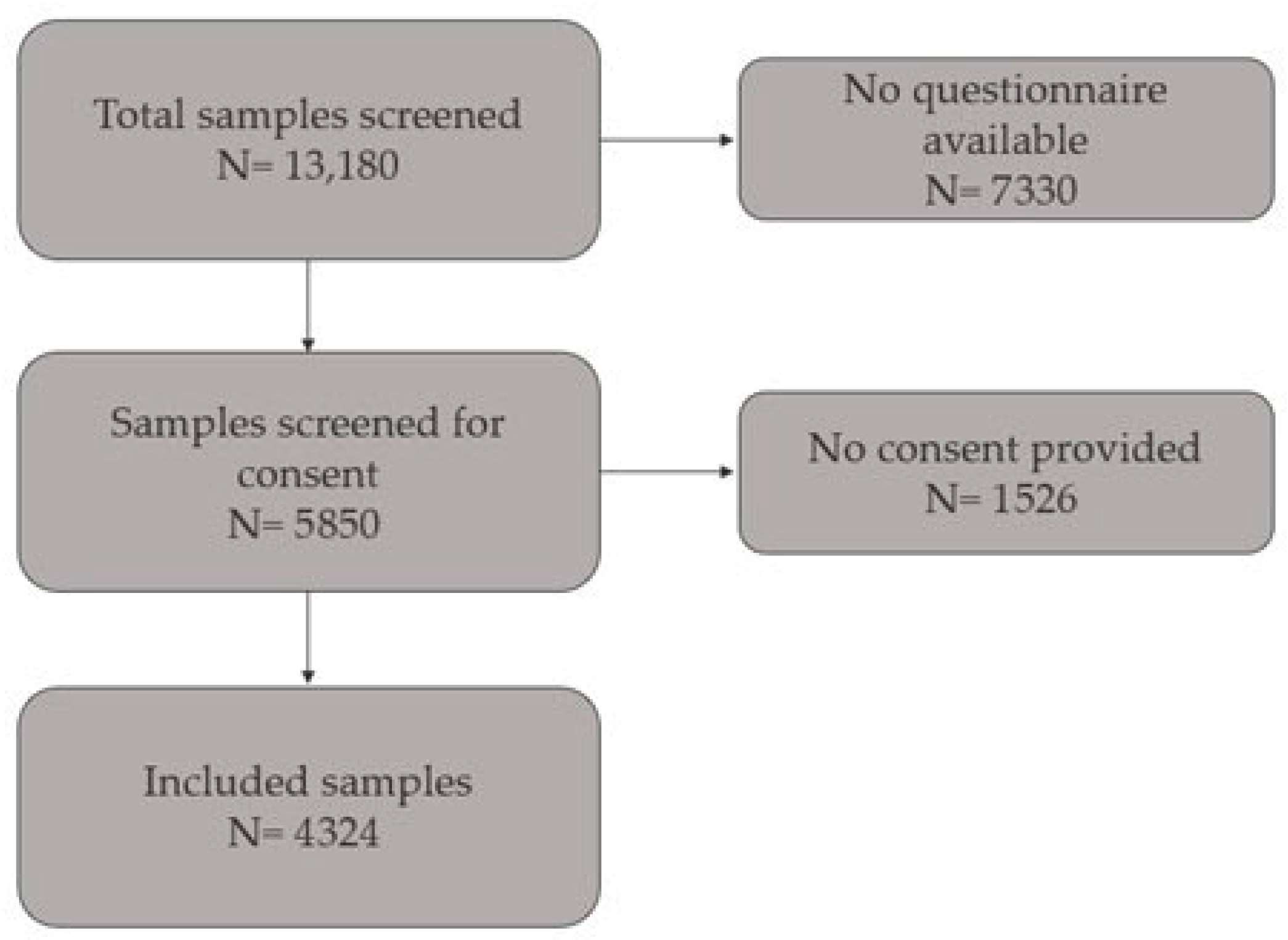
2.1. Study Population
2.2. Phenotypic Classification
2.3. SNP Selection and Genotyping
2.4. Statistical Analysis
3. Results
3.1. Participant Characteristics
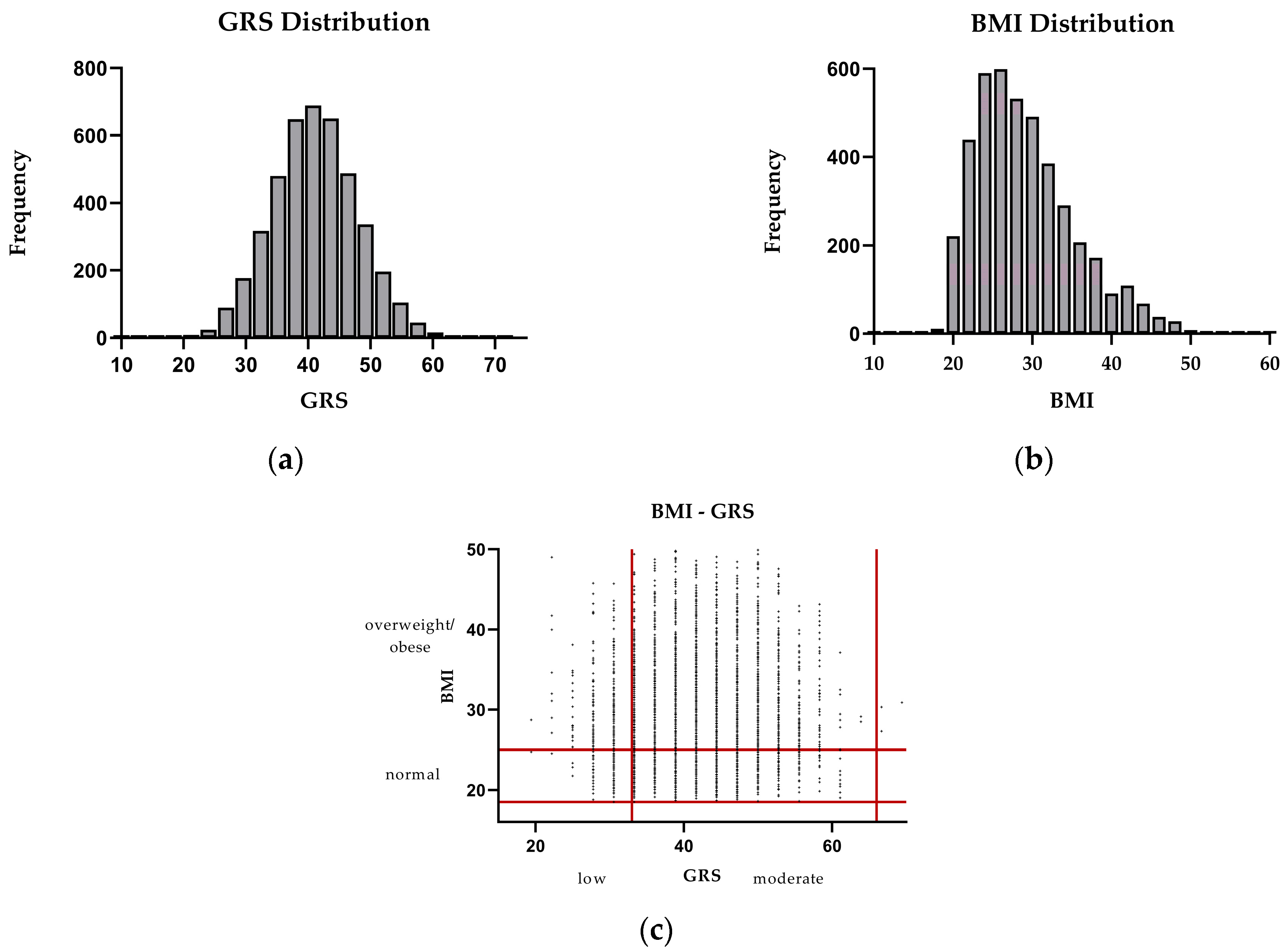
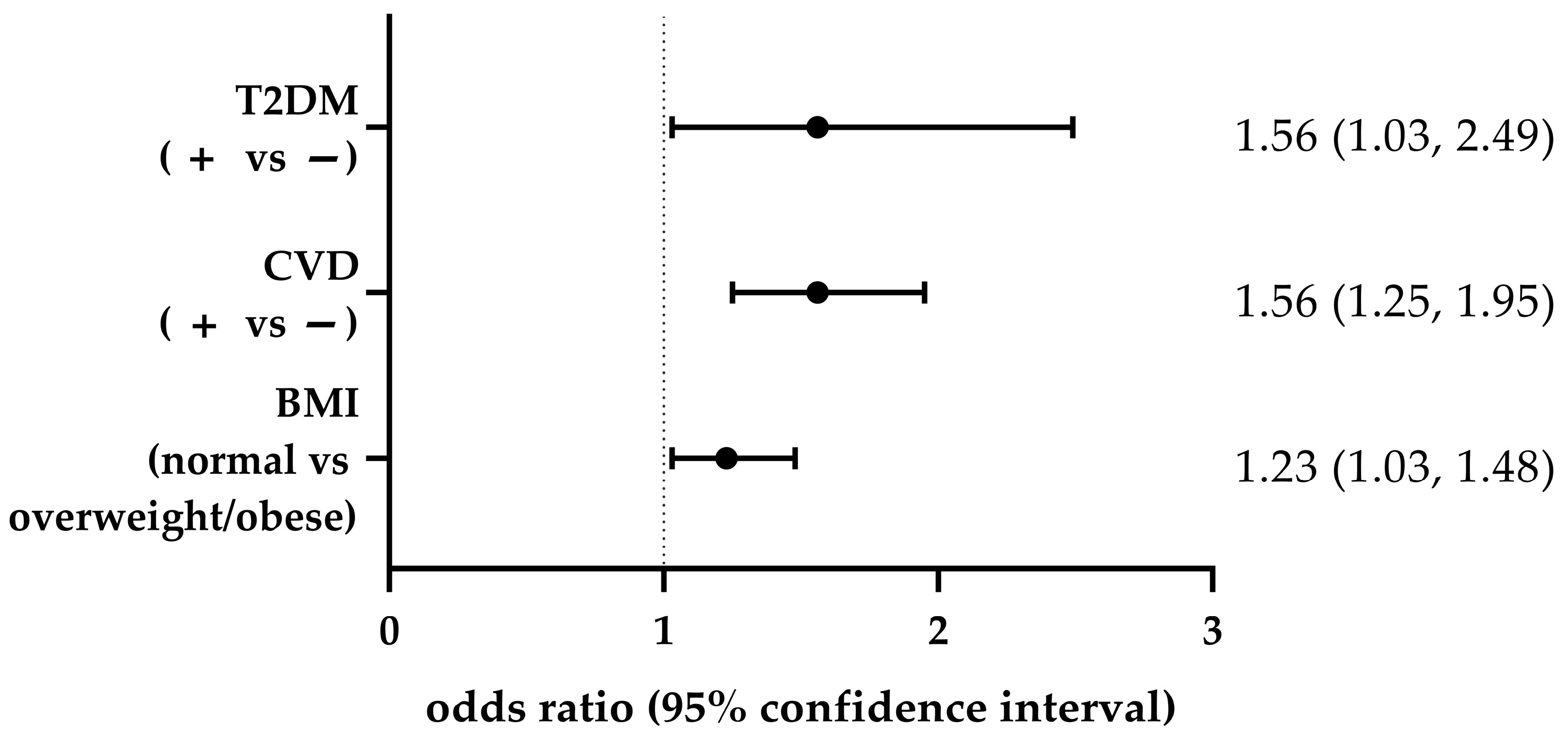
3.2. Associations Between GRS and Obesity, and Between CVD and T2DM
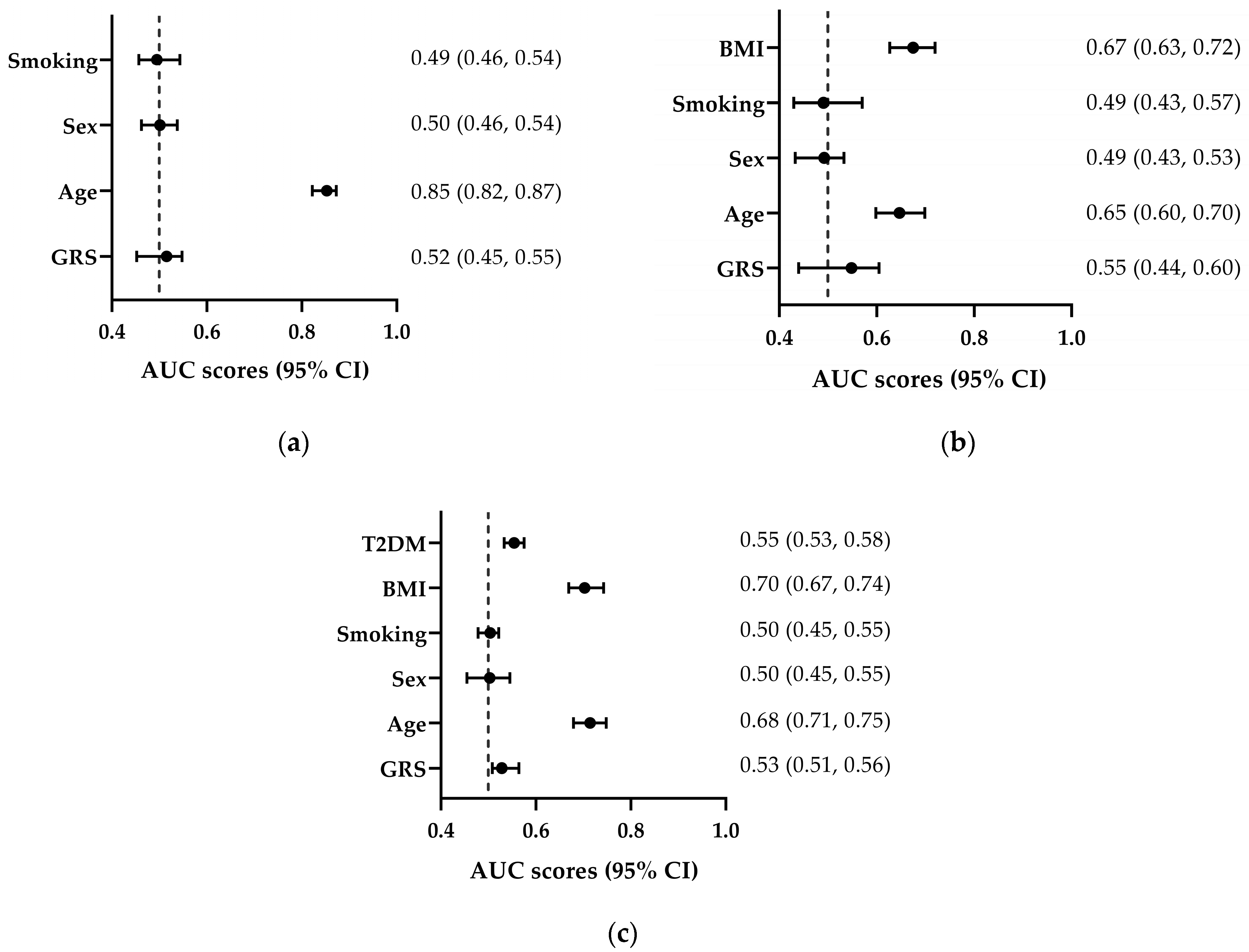
3.3. Discriminative Ability of GRS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE | Angiotensin I Converting Enzyme |
| ADH1C | Alcohol Dehydrogenase 1C (Class I), Gamma Polypeptide |
| ADORA2A | Adenosine A2a Receptor |
| ADRB2 | Adrenoceptor Beta 2 |
| APOC3 | Apolipoprotein C3 |
| APOE | Apolipoprotein E |
| AUC | Area Under the Curve |
| BMI | Body Mass Index |
| CLOCK | Clock Circadian Regulator |
| CVD | Cardiovascular Disease |
| CYP1A2 | Cytochrome P450 Family 1 Subfamily A Member 2 |
| FADS2 | Fatty Acid Desaturase 2 |
| FTO | FTO alpha-ketoglutarate dependent dioxygenase |
| GRS | Genetic Risk Score |
| GWAS | Genome-Wide Association Study |
| MR | Mendelian Randomization |
| LEP | Leptin |
| LEPR | Leptin Receptor |
| LIPC | Hepatic Lipase C |
| MC4R | Melanocortin 4 Receptor |
| MCM6 | Minichromosome Maintenance Complex Component 6 |
| PN | Precision Nutrition |
| POMC | Pro-Opiomelanocortin |
| SLC2A2 | Solute Carrier Family 2 Member 2 (Glucose Transporter Type 2) |
| T2DM | Type 2 Diabetes Mellitus |
| TCF7L2 | Transcription Factor 7 Like 2 |
References
- Wong, N.D.; Sattar, N. Cardiovascular risk in diabetes mellitus: Epidemiology, assessment and prevention. Nat. Rev. Cardiol. 2023, 20, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Verma, A.; Garg, R.; Singh, J.; Verma, H. Cardiometabolic Risk Factors Associated with Type 2 Diabetes Mellitus: A Mechanistic Insight. Clin. Med. Insights Endocrinol. Diabetes 2023, 16, 11795514231220780. [Google Scholar] [CrossRef] [PubMed]
- Koskinas, K.C.; Van Craenenbroeck, E.M.; Antoniades, C.; Blüher, M.; Gorter, T.M.; Hanssen, H.; Marx, N.; McDonagh, T.A.; Mingrone, G.; Rosengren, A.; et al. Obesity and cardiovascular disease: An ESC clinical consensus statement. Eur. Heart J. 2024, 45, 4063–4098. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why Does Obesity Cause Diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. The Role of Obesity in Type 2 Diabetes Mellitus—An Overview. Int. J. Mol. Sci. 2024, 25, 1882. [Google Scholar] [CrossRef] [PubMed]
- World Obesity Federation World Obesity Atlas. 2025. Available online: https://www.worldobesity.org/resources/resource-library/world-obesity-atlas-2025 (accessed on 8 July 2025).
- Powell-Wiley, T.M.; Poirier, C.P.; Burke, V.C.L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef] [PubMed]
- He, K.-J.; Wang, H.; Xu, J.; Gong, G.; Liu, X.; Guan, H. Global burden of type 2 diabetes mellitus from 1990 to 2021, with projections of prevalence to 2044: A systematic analysis across SDI levels for the global burden of disease study 2021. Front. Endocrinol. 2024, 15, 1501690. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Qiu, T.; Li, L.; Yu, R.; Chen, X.; Li, C.; Proud, C.G.; Jiang, T. Pathophysiology of obesity and its associated diseases. Acta Pharm. Sin. B 2023, 13, 2403–2424. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, S.; Zhang, C. The Related Metabolic Diseases and Treatments of Obesity. Healthcare 2022, 10, 1616. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, D.; Vermund, S.H. Advantages and Limitations of the Body Mass Index (BMI) to Assess Adult Obesity. Int. J. Environ. Res. Public Health 2024, 21, 757. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, K.; Garvey, W.T.; Martins, C. Strengths and Limitations of BMI in the Diagnosis of Obesity: What is the Path Forward? Curr. Obes. Rep. 2024, 13, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Shin, S.W. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.K. An Overview of Mongenic and Syndromic Obesities in Humans. Pediatr. Blood Cancer 2012, 58, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Fitch, A.K.; Malhotra, S.; Conroy, R. Differentiating monogenic and syndromic obesities from polygenic obesity: Assessment, diagnosis, and management. Obes. Pillars 2024, 11, 100110. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Genetics of Obesity in Humans: A Clinical Review. Int. J. Mol. Sci. 2022, 23, 11005. [Google Scholar] [CrossRef] [PubMed]
- Wardle, J.; Carnell, S.; Haworth, C.M.; Plomin, R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am. J. Clin. Nutr. 2008, 87, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Penney, K.L.; Giovannucci, E.; Kraft, P.; Wilson, K.M. A genome-wide association study of energy intake and expenditure. PLoS ONE 2018, 13, e0201555. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, F.K.; Huyvaert, M.; Ortalli, A.; Canouil, M.; Lecoeur, C.; Verbanck, M.; Lobbens, S.; Khamis, A.; Marselli, L.; Marchetti, P.; et al. The expression of genes in top obesity-associated loci is enriched in insula and substantia nigra brain regions involved in addiction and reward. Int. J. Obes. 2020, 44, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Duis, J.; Butler, M.G. Syndromic and Nonsyndromic Obesity: Underlying Genetic Causes in Humans. Adv. Biol. 2022, 6, e2101154. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.; Wills, A.K.; Wong, A.; Elks, C.E.; Wareham, N.J.; Loos, R.J.F.; Kuh, D.; Ong, K.K. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum. Mol. Genet. 2010, 19, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Li, L.; Zhang, L.; Guo, L.; Wang, C. Association between MC4R rs17782313 genotype and obesity: A meta-analysis. Gene 2020, 733, 144372. [Google Scholar] [CrossRef] [PubMed]
- Crovesy, L.; Rosado, E.L. Interaction between genes involved in energy intake regulation and diet in obesity. Nutrition 2019, 67–68, 110547. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Elliott, P.; Zabaneh, D.; Zhang, W.; Li, Y.; Froguel, P.; Balding, D.; Scott, J.; Kooner, J.S. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 2008, 40, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Chandak, G.R.; Shen, Y.; Wang, Q.; Zhou, D. Association between Common Polymorphism near the MC4R Gene and Obesity Risk: A Systematic Review and Meta-Analysis. PLoS ONE 2012, 7, e45731. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.A.; Bradfield, J.P.; Zhang, H.; Wang, K.; Kim, C.E.; Annaiah, K.; Santa, E.; Glessner, J.T.; Thomas, K.; Garris, M.; et al. Investigation of the locus near MC4R with childhood obesity in Americans of European and African ancestry. Obesity 2009, 17, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, J.; Wu, J.; Yang, M. Combined effects of FTO rs9939609 and MC4R rs17782313 on elevated nocturnal blood pressure in the Chinese Han population. Cardiovasc. J. Afr. 2016, 27, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Haupt, A.; Preissl, H.; Ketterer, C.; Hennige, A.M.; Sartorius, T.; Machicao, F.; Fritsche, A.; Häring, H.-U. An Obesity Risk SNP (rs17782313) near the MC4R Gene Is Associated with Cerebrocortical Insulin Resistance in Humans. J. Obes. 2011, 2011, 283153. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.K.; Florez, J.C. The genetics of type 2 diabetes: What have we learned from GWAS? Ann. N. Y. Acad. Sci. 2010, 1212, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Aragam, K.G.; Jiang, T.; Goel, A.; Kanoni, S.; Wolford, B.N.; Atri, D.S.; Weeks, E.M.; Wang, M.; Hindy, G.; Zhou, W.; et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022, 54, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Jurgens, S.J.; Erdmann, J.; Bezzina, C.R. Genome-wide association studies of cardiovascular disease. Physiol. Rev. 2023, 103, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Xu, L.; Zhang, L.; Han, Z.; Jiang, Q.; Wang, Z.; Jin, S. Meta-analysis of association between TCF7L2 polymorphism rs7903146 and type 2 diabetes mellitus. BMC Med. Genet. 2018, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- del Bosque-Plata, L.; Martínez-Martínez, E.; Espinoza-Camacho, M.Á.; Gragnoli, C. The Role of TCF7L2 in Type 2 Diabetes. Diabetes 2021, 70, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Toribio-Fernández, R.; Tristão-Pereira, C.; Carlos Silla-Castro, J.; Callejas, S.; Oliva, B.; Fernandez-Nueda, I.; Garcia-Lunar, I.; Perez-Herreras, C.; María Ordovás, J.; Martin, P.; et al. Apolipoprotein E-ε2 and Resistance to Atherosclerosis in Midlife: The PESA Observational Study. Circ. Res. 2024, 134, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Havulinna, A.S.; Bhalala, O.G.; Byars, S.G.; De Livera, A.M.; Yetukuri, L.; Tikkanen, E.; Perola, M.; Schunkert, H.; Sijbrands, E.J.; et al. Genomic prediction of coronary heart disease. Eur. Heart J. 2016, 37, 3267–3278. [Google Scholar] [CrossRef] [PubMed]
- Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genet. 2013, 9, e1003348. [Google Scholar] [CrossRef]
- Zhang, M.; Ward, J.; Strawbridge, R.J.; Celis-Morales, C.; Pell, J.P.; Lyall, D.M.; Ho, F.K. How do lifestyle factors modify the association between genetic predisposition and obesity-related phenotypes? A 4-way decomposition analysis using UK Biobank. BMC Med. 2024, 22, 230. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.; Christensen, S.M.; Richardson, L.; Ingersoll, A.B.; Burridge, K.; Golden, A.; Karjoo, S.; Cortez, D.; Shelver, M.; Bays, H.E. Nutrition and physical activity: An Obesity Medicine Association (OMA) Clinical Practice Statement 2022. Obes. Pillars 2022, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Weir, C.B.; Jan, A. BMI Classification Percentile and Cut Off Points. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M.; et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Mullins, V.A.; Bresette, W.; Johnstone, L.; Hallmark, B.; Chilton, F.H. Genomics in Personalized Nutrition: Can You “Eat for Your Genes”? Nutrients 2020, 12, 3118. [Google Scholar] [CrossRef] [PubMed]
- Singar, S.; Nagpal, R.; Arjmandi, B.H.; Akhavan, N.S. Personalized Nutrition: Tailoring Dietary Recommendations through Genetic Insights. Nutrients 2024, 16, 2673. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, R.L. The Box Plots Alternative for Visualizing Quantitative Data. PM&R 2016, 8, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.M.; Steluti, J.; Norde, M.M.; Fisberg, R.M.; Marchioni, D.M. The association between genetic risk score and blood pressure is modified by coffee consumption: Gene-diet interaction analysis in a population-based study. Clin. Nutr. 2019, 38, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R.; Loos, R.J.F.; O’Rahilly, S.; Hirschhorn, J.N.; Allison, D.B. GWAS for BMI: A treasure trove of fundamental insights into the genetic basis of obesity. Int. J. Obes. 2018, 42, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.B.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A Common Variant in the FTO Gene Is Associated with Body Mass Index and Predisposes to Childhood and Adult Obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.S.; Miranda, N.; Cade, B.E.; Huang, T.; Redline, S.; Karlson, E.W.; Saxena, R. Interaction of obesity polygenic score with lifestyle risk factors in an electronic health record biobank. BMC Med. 2022, 20, 5. [Google Scholar] [CrossRef] [PubMed]
- Ghatan, S.; van Rooij, J.; van Hoek, M.; Boer, C.G.; Felix, J.F.; Kavousi, M.; Jaddoe, V.W.; Sijbrands, E.J.G.; Medina-Gomez, C.; Rivadeneira, F.; et al. Defining type 2 diabetes polygenic risk scores through colocalization and network-based clustering of metabolic trait genetic associations. Genome Med. 2024, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Hubacek, J.A.; Dlouha, L.; Adamkova, V.; Dlouha, D.; Pacal, L.; Kankova, K.; Galuska, D.; Lanska, V.; Veleba, J.; Pelikanova, T. Genetic risk score is associated with T2DM and diabetes complications risks. Gene 2023, 849, 146921. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Wade, K.H.; Zahid, S.; Brancale, J.; Xia, R.; Distefano, M.; Senol-Cosar, O.; Haas, M.E.; Bick, A.; et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 2019, 177, 587–596.e9. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Lazarte, J.; Dron, J.S.; McIntyre, A.D.; Skanes, A.C.; Gula, L.J.; Tang, A.S.; Tadros, R.; Laksman, Z.W.; Hegele, R.A.; Roberts, J.D. Role of Common Genetic Variation in Lone Atrial Fibrillation. Circ. Genom. Precis. Med. 2021, 14, e003179. [Google Scholar] [CrossRef] [PubMed]
- Ntalla, I.; Kanoni, S.; Zeng, L.; Giannakopoulou, O.; Danesh, J.; Watkins, H.; Samani, N.J.; Deloukas, P.; Schunkert, H.; UK Biobank CardioMetabolic Consortium CHD Working Group. Genetic Risk Score for Coronary Disease Identifies Predispositions to Cardiovascular and Noncardiovascular Diseases. J. Am. Coll. Cardiol. 2019, 73, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Sodero, G.; Rigante, D.; Pane, L.C.; Sessa, L.; Quarta, L.; Candelli, M.; Cipolla, C. Cardiometabolic Risk Assessment in a Cohort of Children and Adolescents Diagnosed with Hyperinsulinemia. Diseases 2024, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Damavandi, N.; Soleymaniniya, A.; Bahrami Zadegan, S.; Samiee Aref, M.H.; Zeinali, S. Development of a genetic risk score for obesity predisposition evaluation. Mol. Genet. Genom. 2022, 297, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Belsky, D.W.; Moffitt, T.E.; Sugden, K.; Williams, B.; Houts, R.; McCarthy, J.; Caspi, A. Development and evaluation of a genetic risk score for obesity. Biodemography Soc. Biol. 2013, 59, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lyall, D.M.; Celis-Morales, C.; Ward, J.; Iliodromiti, S.; Anderson, J.J.; Gill, J.M.R.; Smith, D.J.; Ntuk, U.E.; Mackay, D.F.; Holmes, M.V.; et al. Association of Body Mass Index with Cardiometabolic Disease in the UK Biobank: A Mendelian Randomization Study. JAMA Cardiol. 2017, 2, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Henry, A.; Roselli, C.; Lin, H.; Sveinbjörnsson, G.; Fatemifar, G.; Hedman, Å.K.; Wilk, J.B.; Morley, M.P.; Chaffin, M.D.; et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Welsh, A.; Hammad, M.; Piña, I.L.; Kulinski, J. Obesity and cardiovascular health. Eur. J. Prev. Cardiol. 2024, 31, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Stathias, V.; Sotiris, G.R.; Karagiannidis, I.; Bourikas, G.; Martinis, G.; Papazoglou, D.; Tavridou, A.; Papanas, N.; Maltezos, E.; Theodoridis, M.; et al. Exploring genomic structure differences and similarities between the Greek and European HapMap populations: Implications for association studies. Ann. Hum. Genet. 2012, 76, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Drineas, P.; Tsetsos, F.; Plantinga, A.; Lazaridis, I.; Yannaki, E.; Razou, A.; Kanaki, K.; Michalodimitrakis, M.; Perez-Jimenez, F.; De Silvestro, G.; et al. Genetic history of the population of Crete. Ann. Hum. Genet. 2019, 83, 373–388. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | rs Number |
|---|---|
| ACE | rs4343 |
| ADH1C | rs283411 |
| ADORA2A | rs5751876 |
| ADRB2 | rs1042713 |
| APOC3 | rs5128 |
| APOE | rs429358 |
| APOE | rs7412 |
| CLOCK | rs1801260 |
| CYP1A2 | rs762551 |
| FADS2 | rs174570 |
| FTO | rs9930506 |
| FTO | rs9939609 |
| LIPC | rs1800588 |
| MC4R | rs17782313 |
| MCM6 | rs4988235 |
| SLC2A2 | rs5400 |
| TCF7L2 | rs12255372 |
| TCF7L2 | rs7903146 |
| Total (n = 4279) | Low GRS (n = 619) | Intermediate/High GRS (n = 3660) | p | |
|---|---|---|---|---|
| Age 1, years | 41.04 ± 11.61 | 39.60 ± 17.20 | 39.90 ± 17.12 | 0.53 |
| Sex, M/F (%) | 1054 (24.6)/ 3225 (75.4) | 139 (22.5)/ 480 (77.5) | 915 (25.0)/ 2745 (75.0) | 0.19 |
| BMI 1, kg/m2 | 29.09 ± 6.21 | 28.67 ± 6.09 | 29.16 ± 6.22 | 0.06 |
| BMI categories, normal/overweight/obese (%) | 1261 (29.5)/3018 (70.5) | 206 (33.3)/413 (66.7) | 1055 (28.8)/2065 (71.2) | 0.02 |
| T2DM, yes/no (%) | 232 (5.4)/4047 (94.6) | 23 (3.7)/596 (96.3) | 209 (5.7)/3451 (94.3) | 0.04 |
| CVD, yes/no (%) | 1023 (23.9)/3256 (76.1) | 109 (17.6)/510 (82.4) | 914 (25.0)/2746 (75.0) | <0.001 |
| Smoking status, yes/no (%) | 1028 (24.8)/ 3096 (75.1) | 162 (27.2)/ 433 (72.8) | 866 (24.5)/ 2663 (75.5) | 0.16 |
| Genetic Risk Score 1 (GRS) | 41.67 ± 11.11 | 33.33 ± 2.78 | 44.40 ± 8.33 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapellou, A.; Fotis, T.; Vrachnos, D.M.; Salata, E.; Ntoumou, E.; Papailia, S.; Vittas, S. Genetic Variants in Metabolic Pathways and Their Role in Cardiometabolic Risk: An Observational Study of >4000 Individuals. Biomedicines 2025, 13, 1791. https://doi.org/10.3390/biomedicines13081791
Kapellou A, Fotis T, Vrachnos DM, Salata E, Ntoumou E, Papailia S, Vittas S. Genetic Variants in Metabolic Pathways and Their Role in Cardiometabolic Risk: An Observational Study of >4000 Individuals. Biomedicines. 2025; 13(8):1791. https://doi.org/10.3390/biomedicines13081791
Chicago/Turabian StyleKapellou, Angeliki, Thanasis Fotis, Dimitrios Miltiadis Vrachnos, Effie Salata, Eleni Ntoumou, Sevastiani Papailia, and Spiros Vittas. 2025. "Genetic Variants in Metabolic Pathways and Their Role in Cardiometabolic Risk: An Observational Study of >4000 Individuals" Biomedicines 13, no. 8: 1791. https://doi.org/10.3390/biomedicines13081791
APA StyleKapellou, A., Fotis, T., Vrachnos, D. M., Salata, E., Ntoumou, E., Papailia, S., & Vittas, S. (2025). Genetic Variants in Metabolic Pathways and Their Role in Cardiometabolic Risk: An Observational Study of >4000 Individuals. Biomedicines, 13(8), 1791. https://doi.org/10.3390/biomedicines13081791