

Single-Cell Transcriptomic Analysis of Different Liver Fibrosis Models: Elucidating Molecular Distinctions and Commonalities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
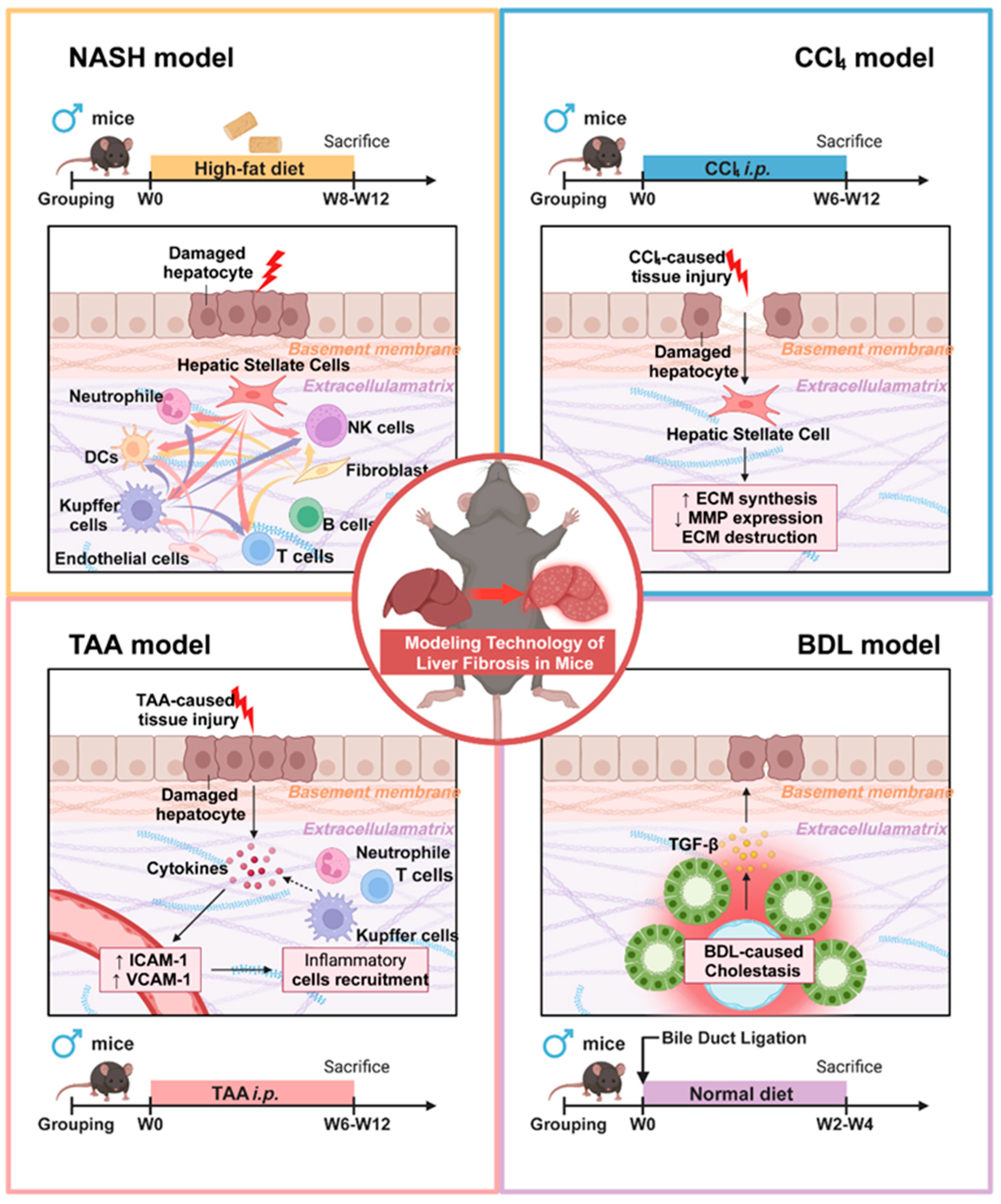
2.2. Animal Models Included
2.3. Data Loading, Quality Control, and Preprocessing
2.4. Dimensionality Reduction and Batch Integration
2.5. Cell Clustering and Visualization
2.6. Cell Type Annotation and Subpopulation Analysis
2.7. Differential Expression and Gene Set Variation Analysis (GSVA)
2.8. Pseudotime Trajectory Analysis
2.9. Cell–Cell Communication Analysis
2.10. Statistical Analysis
3. Results
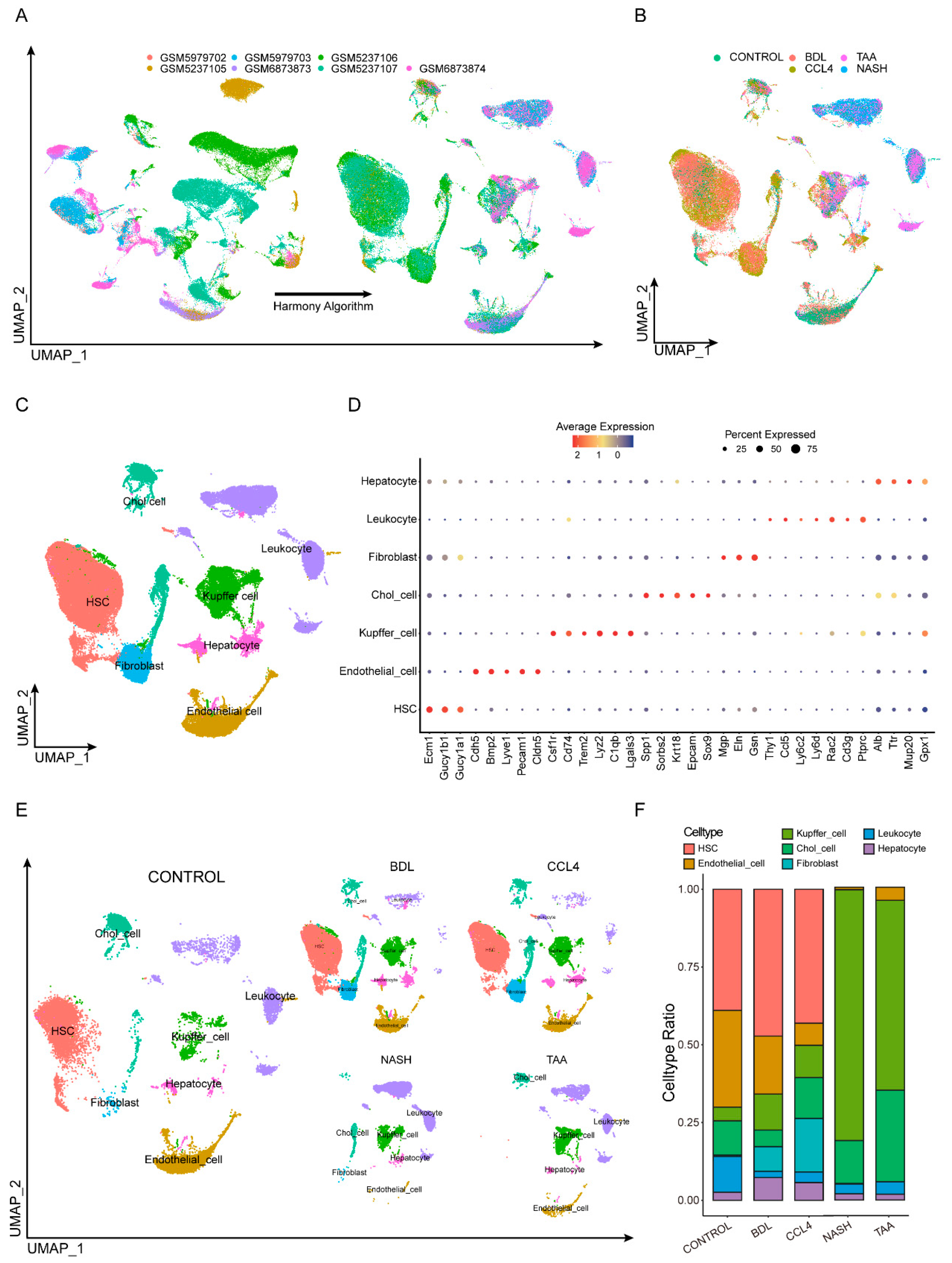
3.1. Integrating Single-Cell Data from Different Modeling Approaches
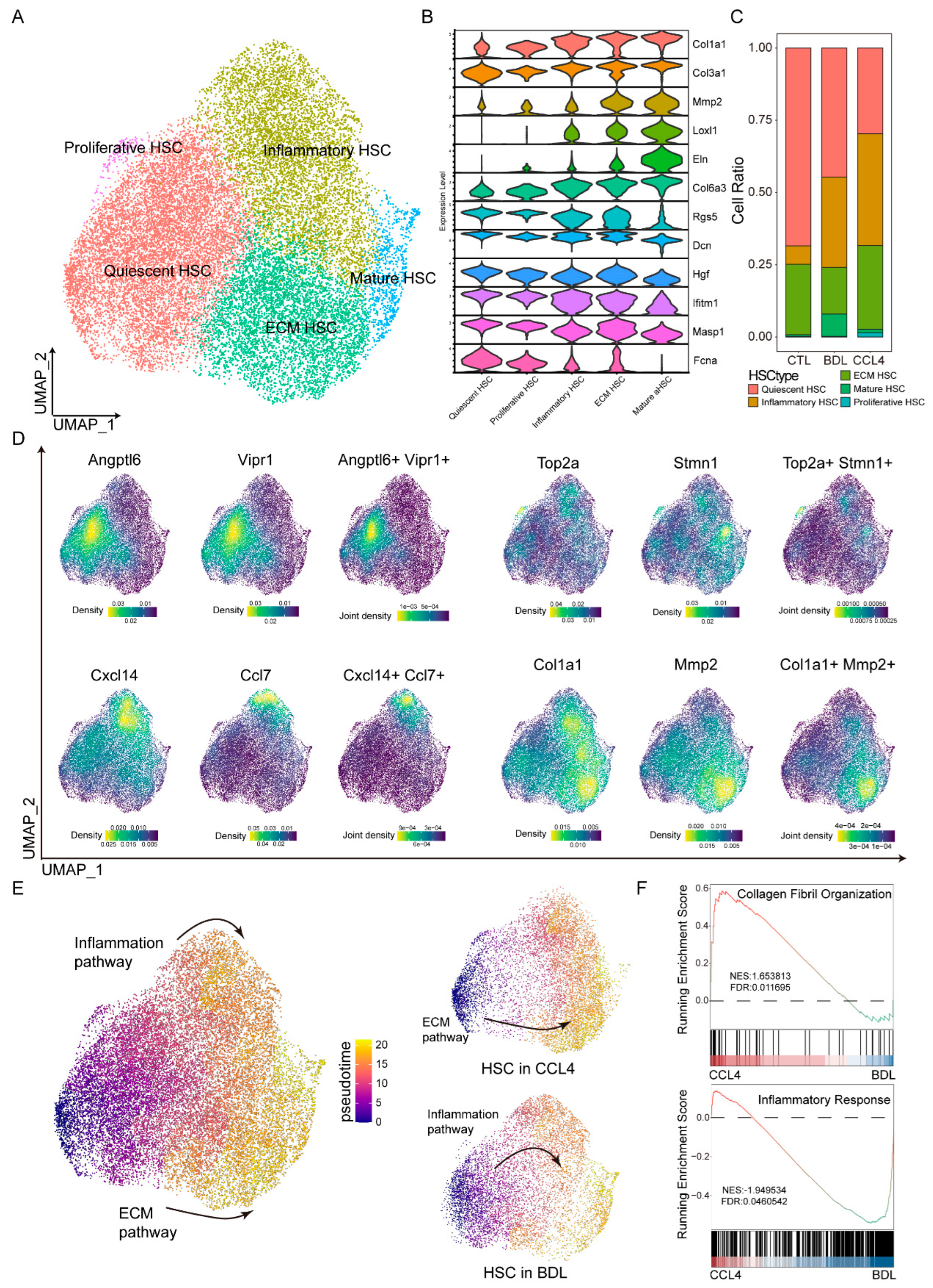
3.2. HSC Differentiation Trajectories Across Different Fibrotic Models
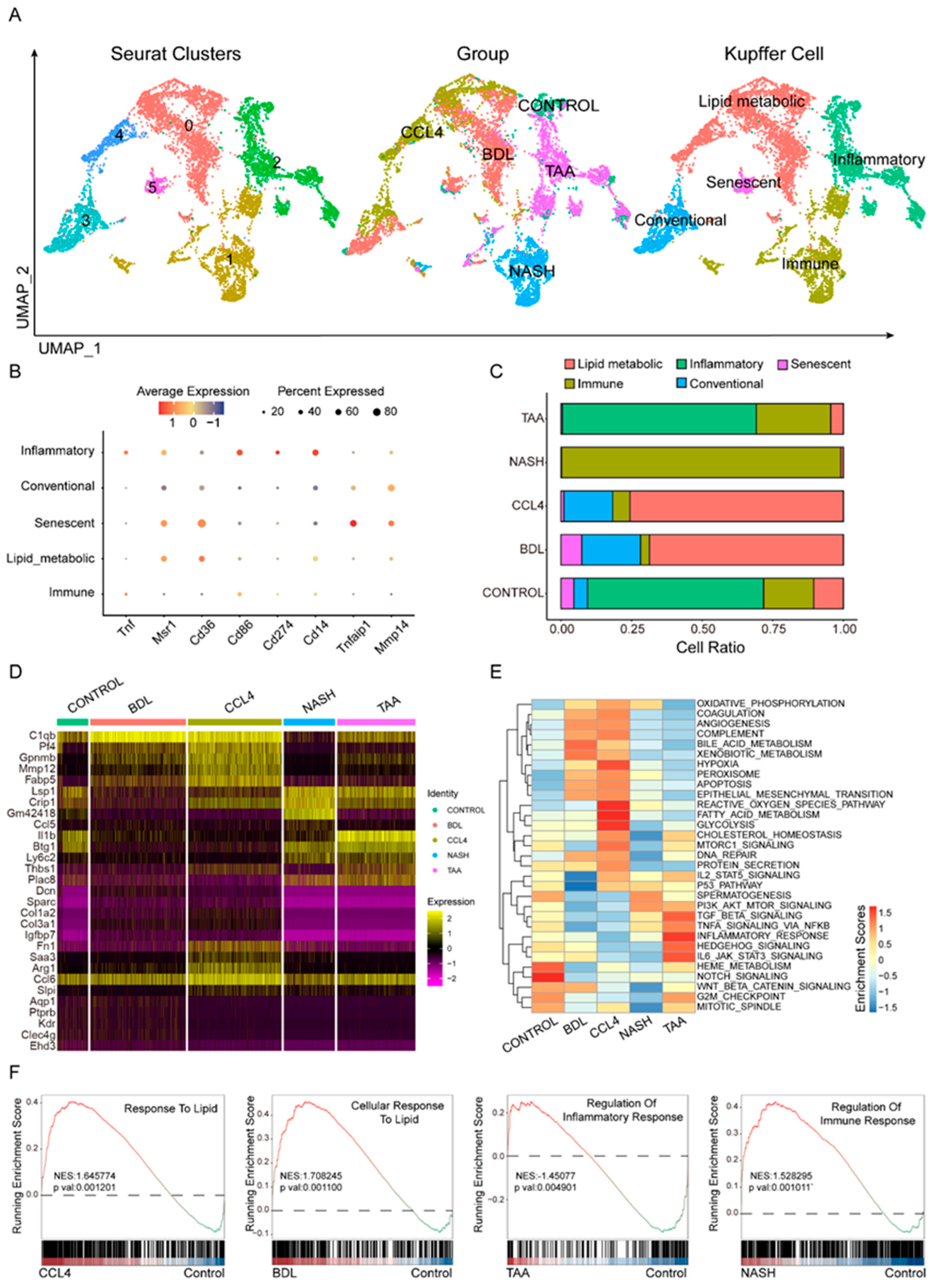
3.3. Heterogeneity of Kupffer Cells in Liver Fibrosis Models
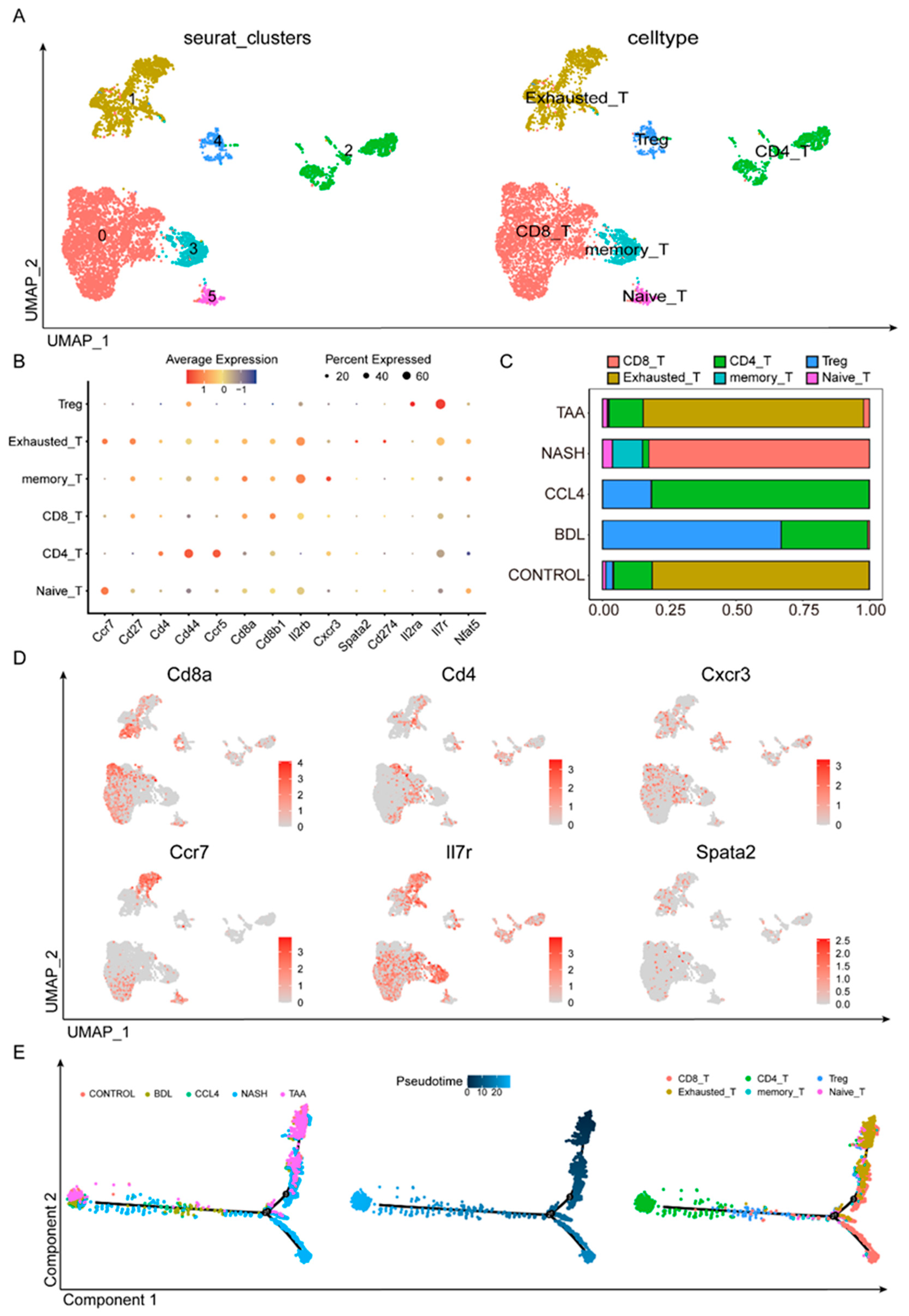
3.4. Dynamic T-Cell Subsets in Liver Fibrosis
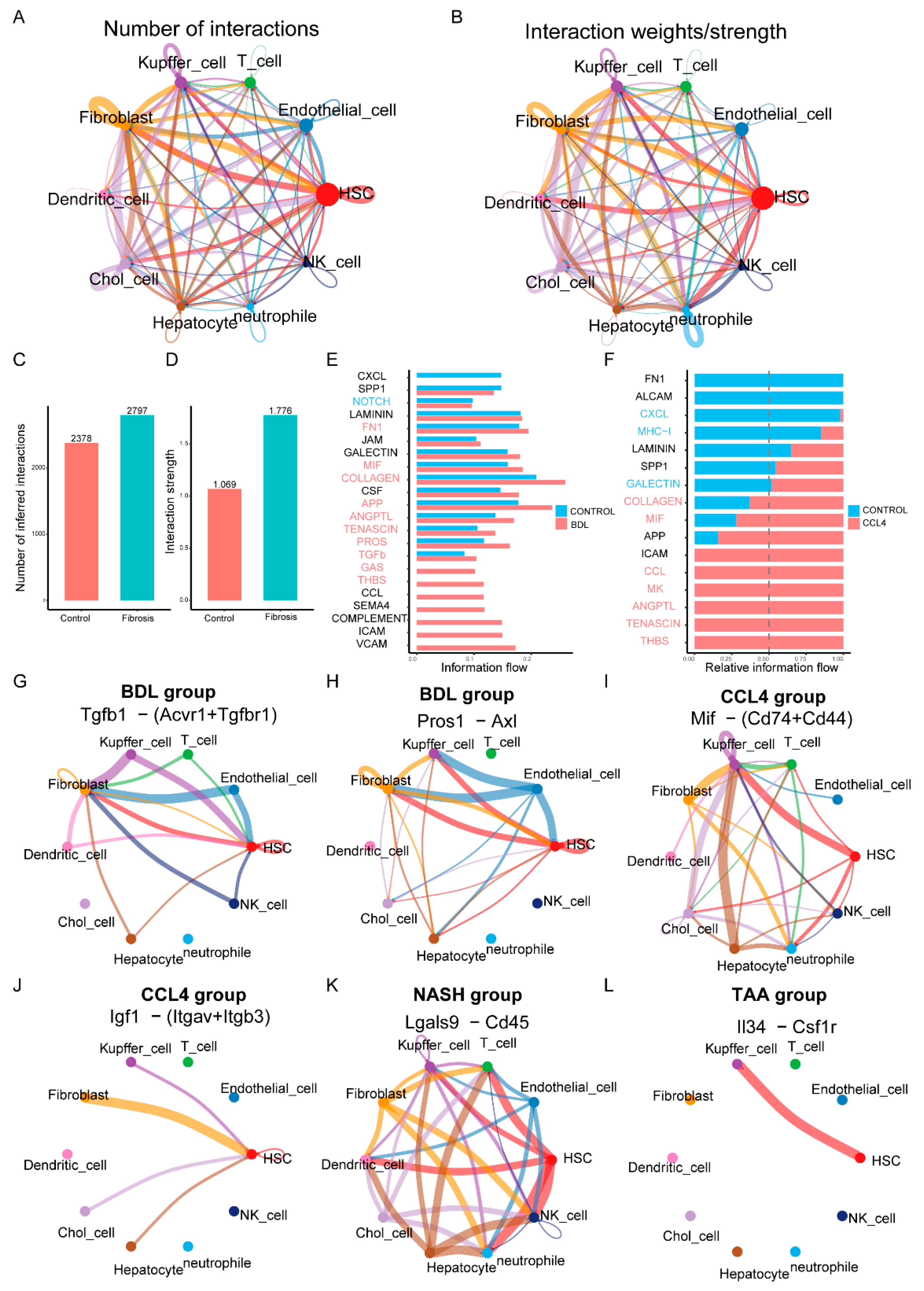
3.5. Cell–Cell Interaction Networks in Liver Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38 (Suppl. S1), S38–S53. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.J.; Lin, Y. Liver Fibrosis Model and Its Construction Method and Application. CN Patent 107988141A, 14 May 2018. [Google Scholar]
- Ding, Z.B.; Gao, Z.; Fu, X.T.; Yao, N.; Chen, J.F.; Li, X.G.; Feng, S.R.; Zhou, J. Method for Constructing a Mouse Spontaneous Liver Cancer Model. CN Patent 116982599A, 3 November 2023. [Google Scholar]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef] [PubMed]
- de Haan, W.; Dheedene, W.; Apelt, K.; Décombas-Deschamps, S.; Vinckier, S.; Verhulst, S.; Conidi, A.; Deffieux, T.; Staring, M.W.; Vandervoort, P.; et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc. Res. 2021, 118, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Zhou, J.C.; Peng, D.; Hua, F.; Li, K.; Yu, J.J.; Lv, X.X.; Cui, B.; Liu, S.S.; Yu, J.M.; et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy 2020, 16, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, T. Animal models for the study of liver fibrosis: New insights from knockout mouse models. Am. J. Physiol. Liver Physiol. 2011, 300, G729–G738. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; He, H.; Wang, T.; Su, N.; Zhang, F.; Jiang, K.; Zhu, J.; Zhang, C.; Niu, K.; Wang, L.; et al. Single-Cell Transcriptomic Analysis Reveals a Hepatic Stellate Cell–Activation Roadmap and Myofibroblast Origin During Liver Fibrosis in Mice. Hepatology 2021, 74, 2774–2790. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.R.; Wong, E.Y.T.; Wong, S.H.; Ng, A.W.T.; Loo, L.H.; Chow, P.K.; Ngeow, J. Global Epidemiology and Genetics of Hepatocellular Carcinoma. Gastroenterology 2023, 164, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nature Reviews. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, C.; Dai, W.; Tong, H.; Yang, W.; Huang, Z.; Tang, C.; Gao, J. Disruption of cholangiocyte-B cell crosstalk by blocking the CXCL12-CXCR4 axis alleviates liver fibrosis. Cell. Mol. Life Sci. 2023, 80, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Motiño, O.; Lambertucci, F.; Chen, H.; Anagnostopoulos, G.; Montégut, L.; Nogueira-Recalde, U.; Maiuri, M.C.; Kroemer, G.; Martins, I. A mouse model of non-alcoholic steatohepatitis and hepatocellular carcinoma induced by Western diet and carbon tetrachloride. Methods Mol. Biol. 2024, 2769, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2022, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Hepatic inflammatory responses in liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Saito, Y.; Nair, A.; Dapito, D.H.; Yu, L.-X.; Ravichandra, A.; Bhattacharjee, S.; Affo, S.; Fujiwara, N.; Su, H.; et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 2022, 610, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; e Rocha, K.C.; Liu, X.; Zhao, H.; Zhang, D.; Jouihan, H.; Pourshahian, S.; Kisseleva, T.; et al. MiR-690 treatment causes decreased fibrosis and steatosis and restores specific Kupffer cell functions in NASH. Cell Metab. 2022, 34, 978–990.e4. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; Wang, G.; Zhang, D.; e Rocha, K.C.; Liu, X.; Zhao, H.; Kisseleva, T.; Brenner, D.A.; et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022, 34, 1201–1213.e5. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Niu, Y.; Liang, K.; Du, Y. Mechanical communication in fibrosis progression. Trends Cell Biol. 2022, 32, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.M.; Iyer, R.; Chakraborty, S. The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability. Cell. Rep. Med. 2023, 4, 101170. [Google Scholar] [CrossRef] [PubMed]
- Barreby, E.; Chen, P.; Aouadi, M. Macrophage functional diversity in NAFLD—More than inflammation. Nat. Rev. Endocrinol. 2022, 18, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Matchett, K.P.; Dobie, R.; Wilson-Kanamori, J.R.; Henderson, N.C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Tacke, F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024, 36, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology 2020, 159, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease–novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, G.; Liang, X.; Pan, Y.; Luo, Y.; Luo, Z.; He, S.; Huang, S.; Chen, Z.; Wang, J.; Fang, S. Single-Cell Transcriptomic Analysis of Different Liver Fibrosis Models: Elucidating Molecular Distinctions and Commonalities. Biomedicines 2025, 13, 1788. https://doi.org/10.3390/biomedicines13081788
Deng G, Liang X, Pan Y, Luo Y, Luo Z, He S, Huang S, Chen Z, Wang J, Fang S. Single-Cell Transcriptomic Analysis of Different Liver Fibrosis Models: Elucidating Molecular Distinctions and Commonalities. Biomedicines. 2025; 13(8):1788. https://doi.org/10.3390/biomedicines13081788
Chicago/Turabian StyleDeng, Guofei, Xiaomei Liang, Yuxi Pan, Yusheng Luo, Zizhen Luo, Shaoxuan He, Shuai Huang, Zhaopeng Chen, Jiancheng Wang, and Shuo Fang. 2025. "Single-Cell Transcriptomic Analysis of Different Liver Fibrosis Models: Elucidating Molecular Distinctions and Commonalities" Biomedicines 13, no. 8: 1788. https://doi.org/10.3390/biomedicines13081788
APA StyleDeng, G., Liang, X., Pan, Y., Luo, Y., Luo, Z., He, S., Huang, S., Chen, Z., Wang, J., & Fang, S. (2025). Single-Cell Transcriptomic Analysis of Different Liver Fibrosis Models: Elucidating Molecular Distinctions and Commonalities. Biomedicines, 13(8), 1788. https://doi.org/10.3390/biomedicines13081788