
Targeting Cellular Senescence: Pathophysiology in Multisystem Age-Related Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cellular Senescence
2.1. Telomere Shortening
2.2. DNA Damage Response
2.3. The Senescence-Associated Secretory Phenotype (SASP)
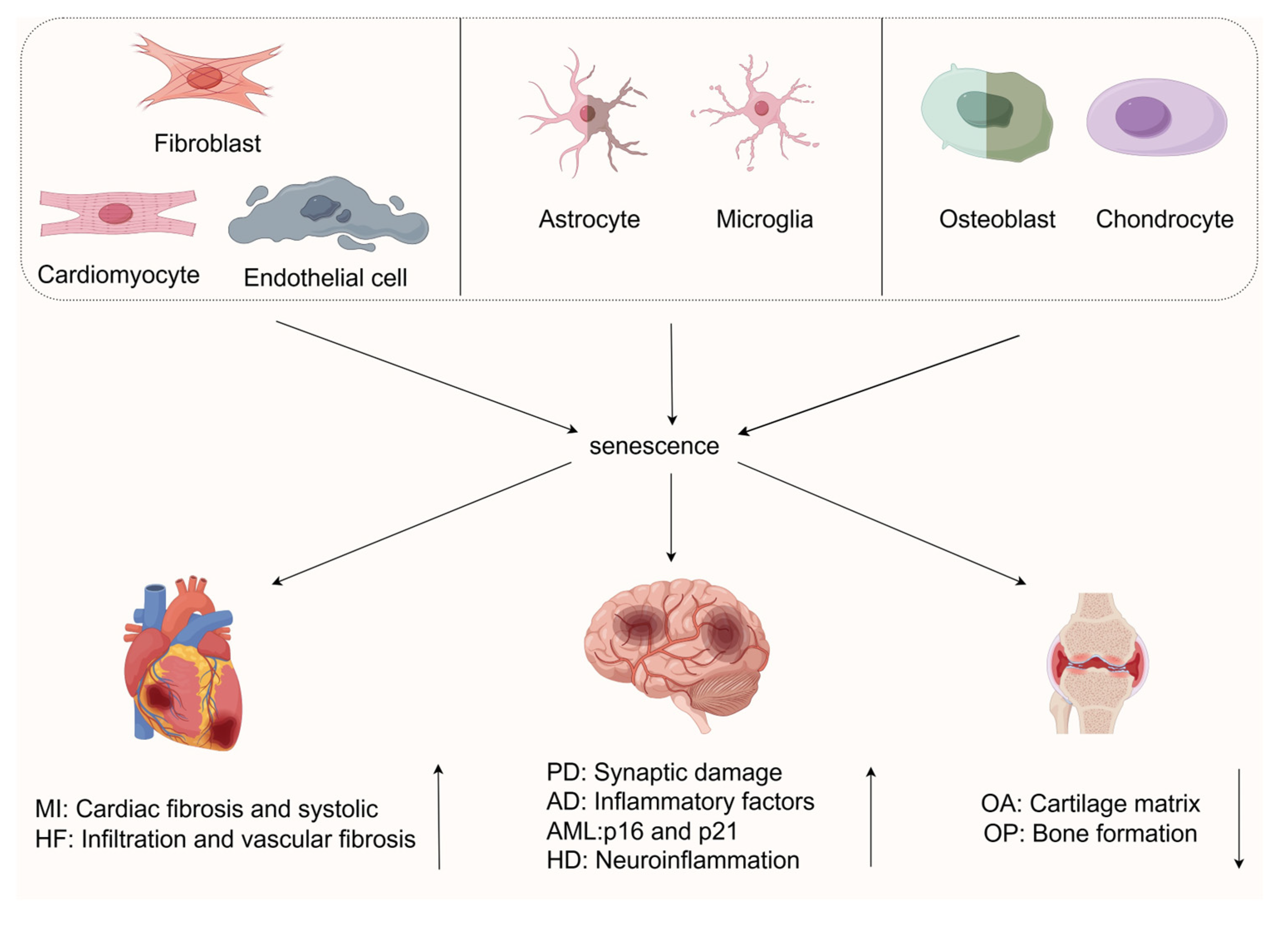
3. Cellular Senescence in Age-Related Diseases
3.1. Cellular Senescence in Cardiovascular Diseases
3.1.1. Cellular Senescence in Myocardial Infarction
3.1.2. Cellular Senescence in Heart Failure
3.1.3. Other Types of Cellular Senescence in Cardiovascular Diseases (CVDs)
3.2. Cellular Senescence in Neurodegenerative Diseases
3.2.1. Cellular Senescence in Alzheimer’s Disease (AD)
Microglia
Astrocytes
3.2.2. Cellular Senescence in Parkinson’s Disease
Microglia
Astrocytes
3.2.3. Cellular Senescence in Other Neurological Disorders
3.3. Cellular Senescence in Musculoskeletal Diseases
3.3.1. Cellular Senescence in Osteoarthritis Inflammation
3.3.2. Cellular Senescence in Osteoporosis
4. Prevention and Treatment of Cellular Senescence
5. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [CrossRef]
- Kroemer, G.; Maier, A.B.; Cuervo, A.M.; Gladyshev, V.N.; Ferrucci, L.; Gorbunova, V.; Kennedy, B.K.; Rando, T.A.; Seluanov, A.; Sierra, F.; et al. From Geroscience to Precision Geromedicine: Understanding and Managing Aging. Cell 2025, 188, 2043–2062. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- de Magalhães, J.P. Cellular Senescence in Normal Physiology. Science 2024, 384, 1300–1301. [Google Scholar] [CrossRef]
- SenNet Recommendations for Detecting Senescent Cells in Different Tissues|Nature Reviews Molecular Cell Biology. Available online: https://www.nature.com/articles/s41580-024-00738-8 (accessed on 23 May 2025).
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Ma, A.; Qu, Y.; Xia, S.; Xu, D.; Ge, C.; Qiu, B.; Xia, Q.; Li, J.; et al. Growth Arrest and DNA Damage 45G Down-Regulation Contributes to Janus Kinase/Signal Transducer and Activator of Transcription 3 Activation and Cellular Senescence Evasion in Hepatocellular Carcinoma. Hepatology 2014, 59, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.; Laspata, N.; Fouquerel, E. Functions of ADP-Ribose Transferases in the Maintenance of Telomere Integrity. Cell Mol. Life Sci. 2022, 79, 215. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-Resolution Fluorescence Imaging of Telomeres Reveals TRF2-Dependent T-Loop Formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Géli, V.; Gilson, E.; Teixeira, M.T. A Two-Step Model for Senescence Triggered by a Single Critically Short Telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Frias, C.; Pampalona, J.; Genesca, A.; Tusell, L. Telomere Dysfunction and Genome Instability. Front. Biosci. (Landmark Ed.) 2012, 17, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sušac, L.; Feigon, J. Structural Biology of Telomerase. Cold Spring Harb. Perspect. Biol. 2019, 11, a032383. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative Stress Shortens Telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Lydall, D. Hiding at the Ends of Yeast Chromosomes: Telomeres, Nucleases and Checkpoint Pathways. J. Cell Sci. 2003, 116, 4057–4065. [Google Scholar] [CrossRef]
- Baird, D.M.; Rowson, J.; Wynford-Thomas, D.; Kipling, D. Extensive Allelic Variation and Ultrashort Telomeres in Senescent Human Cells. Nat. Genet. 2003, 33, 203–207. [Google Scholar] [CrossRef]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21(CIP1), but Not P16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Blackburn, E.H. Telomeres and Telomerase: Their Mechanisms of Action and the Effects of Altering Their Functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors P21 and P16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the Cyclin-Dependent Kinase Inhibitor P16 (INK4a) in Replicative Senescence of Normal Human Fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Ferrell, J.E. A Positive-Feedback-Based Bistable “memory Module” That Governs a Cell Fate Decision. Nature 2003, 426, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, M.; Grekhova, A.; Astrelina, T.; Nikitina, V.; Dobrovolskaya, E.; Suchkova, Y.; Kobzeva, I.; Usupzhanova, D.; Vorobyeva, N.; Samoylov, A.; et al. Accumulation of Spontaneous γH2AX Foci in Long-Term Cultured Mesenchymal Stromal Cells. Aging 2016, 8, 3498–3506. [Google Scholar] [CrossRef]
- Suzuki, M.; Suzuki, K.; Kodama, S.; Yamashita, S.; Watanabe, M. Persistent Amplification of DNA Damage Signal Involved in Replicative Senescence of Normal Human Diploid Fibroblasts. Oxid. Med. Cell Longev. 2012, 2012, 310534. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular Senescence and Its Effector Programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Evangelou, K.; Vasileiou, P.V.S.; Papaspyropoulos, A.; Hazapis, O.; Petty, R.; Demaria, M.; Gorgoulis, V.G. Cellular Senescence and Cardiovascular Diseases: Moving to the “Heart” of the Problem. Physiol. Rev. 2023, 103, 609–647. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by P53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Takasugi, M.; Yoshida, Y.; Ohtani, N. Cellular Senescence and the Tumour Microenvironment. Mol. Oncol. 2022, 16, 3333–3351. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L. Epidemiology of Heart Failure: A Contemporary Perspective. Circ. Res. 2021, 128, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Coussens, L.M.; Stewart, S.A. Senescence and Cancer: An Evolving Inflammatory Paradox. Biochim. Biophys. Acta 2016, 1865, 14–22. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the Ageing Microenvironment Influences Tumour Progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef]
- Wang, X.; Ma, L.; Pei, X.; Wang, H.; Tang, X.; Pei, J.-F.; Ding, Y.-N.; Qu, S.; Wei, Z.-Y.; Wang, H.-Y.; et al. Comprehensive Assessment of Cellular Senescence in the Tumor Microenvironment. Brief. Bioinform. 2022, 23, bbac118. [Google Scholar] [CrossRef]
- Ye, M.; Huang, X.; Wu, Q.; Liu, F. Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices? Cancers 2023, 15, 1927. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Al Suraih, M.S.; Trussoni, C.E.; Splinter, P.L.; LaRusso, N.F.; O’Hara, S.P. Senescent Cholangiocytes Release Extracellular Vesicles That Alter Target Cell Phenotype via the Epidermal Growth Factor Receptor. Liver Int. 2020, 40, 2455–2468. [Google Scholar] [CrossRef]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the Evolution of Cellular Senescence. Aging Cell 2020, 19, e13270. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming in Vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-kappaB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-κB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef]
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.-H.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils Induce Paracrine Telomere Dysfunction and Senescence in ROS-Dependent Manner. EMBO J. 2021, 40, e106048. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An Aged Immune System Drives Senescence and Ageing of Solid Organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Liu, R.-M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (Senescent) Rather than Activated Microglial Cells Are Associated with Tau Pathology and Likely Precede Neurodegeneration in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative Senescence Dictates the Emergence of Disease-Associated Microglia and Contributes to Aβ Pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and Aging: Signaling Pathways and Intervention Therapies. Signal Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable Risk Factors, Cardiovascular Disease, and Mortality in 155722 Individuals from 21 High-Income, Middle-Income, and Low-Income Countries (PURE): A Prospective Cohort Study. Lancet 2020, 395, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The Role of Cellular Senescence in Cardiac Disease: Basic Biology and Clinical Relevance. Nat. Rev. Cardiol. 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Scopus. Nucleolar Expansion and Elevated Protein Translation in Premature Aging. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85028539885&origin=inward&txGid=fc44ef33ee267ab28f4b78b31e7553e3 (accessed on 24 May 2025).
- Scopus. Cellular Senescence: The Good, the Bad and the Unknown. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85135261305&origin=inward&txGid=ca26f9544b701ad679cbf1aa1485ad7a (accessed on 27 June 2025).
- Scopus. Identification of Senescent Cell Surface Targetable Protein DPP4. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85029718809&origin=inward&txGid=9fcbbd29bca55fca89f392d3be66e964 (accessed on 27 June 2025).
- Scopus. Effects of Age-Dependent Changes in Cell Size on Endothelial Cell Proliferation and Senescence Through YAP1s. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85072345071&origin=inward&txGid=511c757186947e5e2413a28f1a820fbb (accessed on 27 June 2025).
- Small Nucleoli Are a Cellular Hallmark of Longevity|Nature Communications. Available online: https://www.nature.com/articles/ncomms16083 (accessed on 27 June 2025).
- Taegtmeyer, H.; Wilson, C.R.; Razeghi, P.; Sharma, S. Metabolic Energetics and Genetics in the Heart. Ann. N. Y. Acad. Sci. 2005, 1047, 208–218. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Pefani, D.-E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA Damage and Protein Stress Responses during Cancer Development and Treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef]
- Programmed Cardiomyocyte Death in Myocardial Infarction|Apoptosis. Available online: https://link.springer.com/article/10.1007/s10495-025-02075-3 (accessed on 28 June 2025).
- Li, D.; Li, Y.; Ding, H.; Wang, Y.; Xie, Y.; Zhang, X. Cellular Senescence in Cardiovascular Diseases: From Pathogenesis to Therapeutic Challenges. J. Cardiovasc. Dev. Dis. 2023, 10, 439. [Google Scholar] [CrossRef]
- Scopus. Cellular Senescence and Organismal Aging. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-45449084795&origin=inward&txGid=48fc2cd49bffef14ca512bdbf5b5e3ea (accessed on 28 June 2025).
- Scopus. Two-Dimensional Design Strategy to Construct Smart Fluorescent Probes for the Precise Tracking of Senescence. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85103187349&origin=inward&txGid=5cd80e54984d7167641600fb720246b1 (accessed on 28 June 2025).
- He, S.; Yan, L.; Yuan, C.; Li, W.; Wu, T.; Chen, S.; Li, N.; Wu, M.; Jiang, J. The Role of Cardiomyocyte Senescence in Cardiovascular Diseases: A Molecular Biology Update. Eur. J. Pharmacol. 2024, 983, 176961. [Google Scholar] [CrossRef]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.-H.; Du, J. Senescent Cardiac Fibroblast Is Critical for Cardiac Fibrosis after Myocardial Infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, W.; Ma, Y.; Chen, B.; Liu, Y.; Piao, C.; Wang, Y.; Yang, M.; Liu, T.; Zhang, J.; et al. Haplodeficiency of Ataxia Telangiectasia Mutated Accelerates Heart Failure after Myocardial Infarction. J. Am. Heart Assoc. 2017, 6, e006349. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.-H.; Chen, H.-Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Cowling, R.T. The Aging Heart, Endothelin-1 and the Senescent Cardiac Fibroblast. J. Mol. Cell Cardiol. 2015, 81, 12–14. [Google Scholar] [CrossRef]
- Scopus. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85037706470&origin=inward&txGid=53f0c213e831998a54aed37e02bf444c (accessed on 28 June 2025).
- Wang, X.; Guo, Z.; Ding, Z.; Khaidakov, M.; Lin, J.; Xu, Z.; Sharma, S.G.; Jiwani, S.; Mehta, J.L. Endothelin-1 Upregulation Mediates Aging-Related Cardiac Fibrosis. J. Mol. Cell Cardiol. 2015, 80, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Meng, Y.; Ding, P.; Wang, H.; Liu, J.; Xia, C.; Chen, Y.; Li, J. Pathological Implication of CaMKII in NF-κB Pathway and SASP during Cardiomyocytes Senescence. Mech. Ageing Dev. 2023, 209, 111758. [Google Scholar] [CrossRef] [PubMed]
- Rouhi, L.; Cheedipudi, S.M.; Chen, S.N.; Fan, S.; Lombardi, R.; Chen, X.; Coarfa, C.; Robertson, M.J.; Gurha, P.; Marian, A.J. Haploinsufficiency of Tmem43 in Cardiac Myocytes Activates the DNA Damage Response Pathway Leading to a Late-Onset Senescence-Associated pro-Fibrotic Cardiomyopathy. Cardiovasc. Res. 2021, 117, 2377–2394. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming Growth Factor (TGF)-β Signaling in Cardiac Remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute Heart Failure. Nat. Rev. Dis. Primers 2020, 6, 16. [Google Scholar] [CrossRef]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting Age-Related Pathways in Heart Failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef]
- Hohensinner, P.J.; Kaun, C.; Buchberger, E.; Ebenbauer, B.; Demyanets, S.; Huk, I.; Eppel, W.; Maurer, G.; Huber, K.; Wojta, J. Age Intrinsic Loss of Telomere Protection via TRF1 Reduction in Endothelial Cells. Biochim. Biophys. Acta 2016, 1863, 360–367. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.Y.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ. Heart Fail. 2017, 10, e003806. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Vita, J.A.; Vasan, R.S.; Levy, D. Changes in Arterial Stiffness and Wave Reflection with Advancing Age in Healthy Men and Women: The Framingham Heart Study. Hypertension 2004, 43, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic Opportunities for Senolysis in Cardiovascular Disease. FEBS J. 2023, 290, 1235–1255. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.K.; Bennett, M.R. Ageing Induced Vascular Smooth Muscle Cell Senescence in Atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short-Term Sustained Hyperglycaemia Fosters an Archetypal Senescence-Associated Secretory Phenotype in Endothelial Cells and Macrophages. Redox Biol. 2018, 15, 170–181. [Google Scholar] [CrossRef]
- Matacchione, G.; Gurău, F.; Silvestrini, A.; Tiboni, M.; Mancini, L.; Valli, D.; Rippo, M.R.; Recchioni, R.; Marcheselli, F.; Carnevali, O.; et al. Anti-SASP and Anti-Inflammatory Activity of Resveratrol, Curcumin and β-Caryophyllene Association on Human Endothelial and Monocytic Cells. Biogerontology 2021, 22, 297–313. [Google Scholar] [CrossRef]
- Inuzuka, Y.; Okuda, J.; Kawashima, T.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Yoshida, Y.; Kosugi, R.; Watanabe-Maeda, K.; et al. Suppression of Phosphoinositide 3-Kinase Prevents Cardiac Aging in Mice. Circulation 2009, 120, 1695–1703. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Geng, Y.-J.; Liang, J.-L.; Zhang, S.; Lei, H.-P.; Zhong, S.-L.; Lin, Q.-X.; Shan, Z.-X.; Lin, S.-G.; Li, Y. High Levels of Glucose Induce “Metabolic Memory” in Cardiomyocyte via Epigenetic Histone H3 Lysine 9 Methylation. Mol. Biol. Rep. 2012, 39, 8891–8898. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Zhao, Y.; Lu, X. Cellular Senescence as a Key Player in Chronic Heart Failure Pathogenesis: Unraveling Mechanisms and Therapeutic Opportunities. Prog. Biophys. Mol. Biol. 2025, 196, 8–18. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-Senescent Cells Contribute to Impaired Heart Regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Smith, A.J.; Lewis, F.C.; Aquila, I.; Waring, C.D.; Nocera, A.; Agosti, V.; Nadal-Ginard, B.; Torella, D.; Ellison, G.M. Isolation and Characterization of Resident Endogenous C-Kit+ Cardiac Stem Cells from the Adult Mouse and Rat Heart. Nat. Protoc. 2014, 9, 1662–1681. [Google Scholar] [CrossRef]
- Scopus. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85042944408&origin=inward&txGid=9922e6f43e60fd3f4b1d97b280bb46ab (accessed on 29 June 2025).
- Uryga, A.K.; Grootaert, M.O.J.; Garrido, A.M.; Oc, S.; Foote, K.; Chappell, J.; Finigan, A.; Rossiello, F.; d’Adda di Fagagna, F.; Aravani, D.; et al. Telomere Damage Promotes Vascular Smooth Muscle Cell Senescence and Immune Cell Recruitment after Vessel Injury. Commun. Biol. 2021, 4, 611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Li, D.-J.; Jiang, Y.-J.; Tong, J.; Fu, H.; Wu, Y.-H.; Shen, F.-M. Vascular Smooth Muscle Cell Senescence and Age-Related Diseases: State of the Art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Scopus. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85126624223&origin=inward&txGid=ef804c76b6801fceeaa4bd6c46e9c8b7 (accessed on 29 June 2025).
- Scopus. Effect of Irbesartan on AGEs-RAGE and MMPs Systems in Rat Type 2 Diabetes Myocardial-Fibrosis Model. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85065249896&origin=inward&txGid=e84f54d3b5057857e4bcf7322fb386d4 (accessed on 29 June 2025).
- Scopus. Neurogenesis in Neurodegenerative Diseases: Role of mfg-e8. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85068549364&origin=inward&txGid=725315541adfdc5745e49663cb7f7db7 (accessed on 29 June 2025).
- Culig, L.; Chu, X.; Bohr, V.A. Neurogenesis in Aging and Age-Related Neurodegenerative Diseases. Ageing Res. Rev. 2022, 78, 101636. [Google Scholar] [CrossRef]
- Wang, Y.; Kuca, K.; You, L.; Nepovimova, E.; Heger, Z.; Valko, M.; Adam, V.; Wu, Q.; Jomova, K. The Role of Cellular Senescence in Neurodegenerative Diseases. Arch. Toxicol. 2024, 98, 2393–2408. [Google Scholar] [CrossRef]
- Neuronal Senescence in the Aged Brain. Available online: https://www.aginganddisease.org/EN/10.14336/AD.2023.0214 (accessed on 29 June 2025).
- Neuronal Senescence May Drive Brain Aging|Science. Available online: https://www.science.org/doi/10.1126/science.adi3450 (accessed on 29 June 2025).
- Saito, Y.; Yamamoto, S.; Chikenji, T.S. Role of Cellular Senescence in Inflammation and Regeneration. Inflamm. Regen. 2024, 44, 28. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular Mechanisms of Neurodegeneration in Alzheimer’s Disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Shang, D.; Hong, Y.; Xie, W.; Tu, Z.; Xu, J. Interleukin-1β Drives Cellular Senescence of Rat Astrocytes Induced by Oligomerized Amyloid β Peptide and Oxidative Stress. Front. Neurol. 2020, 11, 929. [Google Scholar] [CrossRef]
- The Role of Atypical Chemokine Receptors in Neuroinflammation and Neurodegenerative Disorders. Available online: https://www.mdpi.com/1422-0067/24/22/16493#B55-ijms-24-16493 (accessed on 29 June 2025).
- Chien, H.-T.; Li, C.-Y.; Su, W.-H.; Chang, K.-C.; Chen, C.-S.; Liu, Y.-T.; Chen, C.-Y.; Dai, C.-Y.; Wang, S.-C. Multi-Omics Profiling of Chemotactic Characteristics of Brain Microglia and Astrocytoma. Life Sci. 2023, 330, 121855. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Boccardi, V.; Pelini, L.; Ercolani, S.; Ruggiero, C.; Mecocci, P. From Cellular Senescence to Alzheimer’s Disease: The Role of Telomere Shortening. Ageing Res. Rev. 2015, 22, 1–8. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia Derived from Aging Mice Exhibit an Altered Inflammatory Profile. GLIA 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Vogels, T.; Murgoci, A.-N.; Hromádka, T. Intersection of Pathological Tau and Microglia at the Synapse. Acta Neuropathol. Commun. 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Astrocytic Metabolic and Inflammatory Changes as a Function of Age—Jiang—2014—Aging Cell—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1111/acel.12268 (accessed on 29 June 2025).
- Pekny, M.; Nilsson, M. Astrocyte Activation and Reactive Gliosis. GLIA 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Cellular Senescence and Neurodegeneration|Human Genetics. Available online: https://link.springer.com/article/10.1007/s00439-023-02565-x (accessed on 29 June 2025).
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, Neuroinflammation and Biological Remodeling as Key Factors in Pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Herrero, M.-T.; Estrada, C.; Maatouk, L.; Vyas, S. Inflammation in Parkinson’s Disease: Role of Glucocorticoids. Front. Neuroanat. 2015, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Glial Pathology but Absence of Apoptotic Nigral Neurons in Long-Standing Parkinson’s Disease—Banati—1998—Movement Disorders—Wiley Online Library. Available online: https://movementdisorders.onlinelibrary.wiley.com/doi/10.1002/mds.870130205 (accessed on 29 June 2025).
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated α-Synuclein Induces the Loss of Dopaminergic Neurons in the Substantia Nigra of Rats. PLoS ONE 2010, 5, e9956. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0009956 (accessed on 29 June 2025). [CrossRef] [PubMed]
- Bianchi, R.; Adami, C.; Giambanco, I.; Donato, R. S100B Binding to RAGE in Microglia Stimulates COX-2 Expression. J. Leukoc. Biol. 2007, 81, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Scopus. Deficiency of Inducible Nitric Oxide Synthase Protects Against MPTP Toxicity In Vivo. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-0034006478&origin=inward&txGid=dbb5f6356bde0c4ba7cea6df098cc571 (accessed on 29 June 2025).
- Scopus. Direct Transfer of α-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-77950571596&origin=inward&txGid=f10b3f19286caa94119104fca994d266 (accessed on 29 June 2025).
- Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef]
- Han, X.; Lei, Q.; Xie, J.; Liu, H.; Li, J.; Zhang, X.; Zhang, T.; Gou, X. Potential Regulators of the Senescence-Associated Secretory Phenotype During Senescence and Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 2207–2218. [Google Scholar] [CrossRef]
- Expression of P16 and P21 in the Frontal Association Cortex of ALS/MND Brains Suggests Neuronal Cell Cycle Dysregulation and Astrocyte Senescence in Early Stages of the Disease—Vazquez-Villaseñor—2020—Neuropathology and Applied Neurobiology—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1111/nan.12559 (accessed on 29 June 2025).
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential Disease-Modifying Therapies for Huntington’s Disease: Lessons Learned and Future Opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef]
- Trias, E.; Beilby, P.R.; Kovacs, M.; Ibarburu, S.; Varela, V.; Barreto-Núñez, R.; Bradford, S.C.; Beckman, J.S.; Barbeito, L. Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS. Front. Aging Neurosci. 2019, 11, 42. [Google Scholar] [CrossRef]
- Chandra, A.; Rajawat, J. Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. Int. J. Mol. Sci. 2021, 22, 3553. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.-A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, Regional and National Burden of Osteoarthritis 1990–2017: A Systematic Analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef]
- Aibar-Almazán, A.; Voltes-Martínez, A.; Castellote-Caballero, Y.; Afanador-Restrepo, D.F.; Carcelén-Fraile, M.D.C.; López-Ruiz, E. Current Status of the Diagnosis and Management of Osteoporosis. Int. J. Mol. Sci. 2022, 23, 9465. [Google Scholar] [CrossRef]
- Jang, S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef]
- He, X.; Hu, W.; Zhang, Y.; Chen, M.; Ding, Y.; Yang, H.; He, F.; Gu, Q.; Shi, Q. Cellular Senescence in Skeletal Disease: Mechanisms and Treatment. Cell Mol. Biol. Lett. 2023, 28, 88. [Google Scholar] [CrossRef]
- Cartilage Homeostasis and Osteoarthritis—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35682994/ (accessed on 29 June 2025).
- Lotz, M.; Loeser, R.F. Effects of Aging on Articular Cartilage Homeostasis. Bone 2012, 51, 241–248. [Google Scholar] [CrossRef]
- Li, Y.; Hou, C.-L.; Ma, X.-R.; Zhong, B.-L.; Zang, Y.; Jia, F.-J.; Lin, Y.-Q.; Lai, K.Y.C.; Chiu, H.F.K.; Ungvari, G.S.; et al. Quality of Life in Chinese Patients with Schizophrenia Treated in Primary Care. Psychiatry Res. 2017, 254, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent Cells and Osteoarthritis: A Painful Connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Xie, J.; Huang, Z.; Yu, X.; Zhou, L.; Pei, F. Clinical Implications of Macrophage Dysfunction in the Development of Osteoarthritis of the Knee. Cytokine Growth Factor. Rev. 2019, 46, 36–44. [Google Scholar] [CrossRef]
- Poudel, S.B.; Ruff, R.R.; Yildirim, G.; Miller, R.A.; Harrison, D.E.; Strong, R.; Kirsch, T.; Yakar, S. Development of Primary Osteoarthritis during Aging in Genetically Diverse UM-HET3 Mice. Res. Sq. 2024, 26, rs.3.rs-3858256. [Google Scholar] [CrossRef]
- Tsuchida, A.I.; Beekhuizen, M.; ’t Hart, M.C.; Radstake, T.R.D.J.; Dhert, W.J.A.; Saris, D.B.F.; van Osch, G.J.V.M.; Creemers, L.B. Cytokine Profiles in the Joint Depend on Pathology, but Are Different between Synovial Fluid, Cartilage Tissue and Cultured Chondrocytes. Arthritis Res. Ther. 2014, 16, 441. [Google Scholar] [CrossRef]
- Falvino, A.; Gasperini, B.; Cariati, I.; Bonanni, R.; Chiavoghilefu, A.; Gasbarra, E.; Botta, A.; Tancredi, V.; Tarantino, U. Cellular Senescence: The Driving Force of Musculoskeletal Diseases. Biomedicines 2024, 12, 1948. [Google Scholar] [CrossRef]
- Scopus. Targeting the Senescence-Related Genes MAPK12 and FOS to Alleviate Osteoarthritis. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85196263402&origin=inward&txGid=ebcaf86e99528019635ae9b177a08a14 (accessed on 29 June 2025).
- Foessl, I.; Dimai, H.P.; Obermayer-Pietsch, B. Long-Term and Sequential Treatment for Osteoporosis. Nat. Rev. Endocrinol. 2023, 19, 520–533. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Law, S.F.; Chandra, A.; Pignolo, R.J. An Unbiased Proteomics Approach to Identify the Senescence-Associated Secretory Phenotype of Human Bone Marrow-Derived Mesenchymal Stem Cells. Bone Rep. 2023, 18, 101674. [Google Scholar] [CrossRef]
- Saul, D.; Khosla, S. Fracture Healing in the Setting of Endocrine Diseases, Aging, and Cellular Senescence. Endocr. Rev. 2022, 43, 984–1002. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Lin, X.; Boyce, B.F.; Zhang, H.; Xing, L. Age-Associated Callus Senescent Cells Produce TGF-Β1 That Inhibits Fracture Healing in Aged Mice. J. Clin. Investig. 2022, 132, e148073. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA Damage and Senescence in Osteoprogenitors Expressing Osx1 May Cause Their Decrease with Age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Scopus. Bone Aging, Cellular Senescence, and Osteoporosis. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85103927681&origin=inward&txGid=1fdfdfa2f7e6798426027137bfb54a3a (accessed on 29 June 2025).
- Targeting Cell Senescence for the Treatment of Age-Related Bone Loss|Current Osteoporosis Reports. Available online: https://link.springer.com/article/10.1007/s11914-019-00504-2 (accessed on 29 June 2025).
- Scopus. Local Senolysis in Aged Mice Only Partially Replicates the Benefits of Systemic Senolysis. Available online: https://www.scopus.com/record/display.uri?eid=2-s2.0-85152618606&origin=inward&txGid=05b37954c452c773521bb41f8327b8ee (accessed on 29 June 2025).
- Chen, X.-K.; Yi, Z.-N.; Wong, G.T.-C.; Hasan, K.M.M.; Kwan, J.S.-K.; Ma, A.C.-H.; Chang, R.C.-C. Is Exercise a Senolytic Medicine? A Systematic Review. Aging Cell 2021, 20, e13294. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, H.; Xiao, N.; Liang, G.; Lin, Y.; Yang, X.; Yang, J.; Qian, Z.; Fu, Y.; Zhang, C.; et al. Aging and Lifestyle Modifications for Preventing Aging-Related Diseases. FASEB J. 2025, 39, e70575. [Google Scholar] [CrossRef]
- Garatachea, N.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Santos-Lozano, A.; Fiuza-Luces, C.; Morán, M.; Emanuele, E.; Joyner, M.J.; Lucia, A. Exercise Attenuates the Major Hallmarks of Aging. Rejuvenation Res. 2015, 18, 57–89. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, W.; Zeng, L.-Q.; Bai, H.; Li, J.; Zhou, J.; Zhou, G.-Y.; Fang, C.-W.; Wang, F.; Qin, X.-J. Exercise and Dietary Intervention Ameliorate High-Fat Diet-Induced NAFLD and Liver Aging by Inducing Lipophagy. Redox Biol. 2020, 36, 101635. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric Restriction Improves Health and Survival of Rhesus Monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Vera, E.; Bernardes de Jesus, B.; Foronda, M.; Flores, J.M.; Blasco, M.A. Telomerase Reverse Transcriptase Synergizes with Calorie Restriction to Increase Health Span and Extend Mouse Longevity. PLoS ONE 2013, 8, e53760. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Pak, H.H.; Haws, S.A.; Green, C.L.; Koller, M.; Lavarias, M.T.; Richardson, N.E.; Yang, S.E.; Dumas, S.N.; Sonsalla, M.; Bray, L.; et al. Fasting Drives the Metabolic, Molecular and Geroprotective Effects of a Calorie-Restricted Diet in Mice. Nat. Metab. 2021, 3, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, S.; Geng, L.; Song, M.; Wang, W.; Ye, Y.; Ji, Q.; Zou, Z.; Wang, S.; He, X.; et al. Caloric Restriction Reprograms the Single-Cell Transcriptional Landscape of Rattus Norvegicus Aging. Cell 2020, 180, 984–1001.e22. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The Effect of Fasting or Calorie Restriction on Autophagy Induction: A Review of the Literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, J.; Zeng, H.; Shao, L. Senescent Cell Depletion Through Targeting BCL-Family Proteins and Mitochondria. Front. Physiol. 2020, 11, 593630. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Yu, Y.; Ren, J.; Liu, Q.; Bao, Z.; Sun, S.; Liu, X.; Ma, S.; Liu, Z.; et al. Nuclear Lamina Erosion-Induced Resurrection of Endogenous Retroviruses Underlies Neuronal Aging. Cell Rep. 2023, 42, 113396. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting Cellular Senescence with Senotherapeutics: Senolytics and Senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yu, H.; Xu, Y. Targeting Cellular Senescence: Pathophysiology in Multisystem Age-Related Diseases. Biomedicines 2025, 13, 1727. https://doi.org/10.3390/biomedicines13071727
Liu J, Yu H, Xu Y. Targeting Cellular Senescence: Pathophysiology in Multisystem Age-Related Diseases. Biomedicines. 2025; 13(7):1727. https://doi.org/10.3390/biomedicines13071727
Chicago/Turabian StyleLiu, Jinxue, Hongliang Yu, and Yuanyuan Xu. 2025. "Targeting Cellular Senescence: Pathophysiology in Multisystem Age-Related Diseases" Biomedicines 13, no. 7: 1727. https://doi.org/10.3390/biomedicines13071727
APA StyleLiu, J., Yu, H., & Xu, Y. (2025). Targeting Cellular Senescence: Pathophysiology in Multisystem Age-Related Diseases. Biomedicines, 13(7), 1727. https://doi.org/10.3390/biomedicines13071727