
The Role of Human Endogenous Retroviruses in the Initiation and Progression of Melanoma

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Epidemiology, Pathogenesis, and Current Treatment of Melanoma
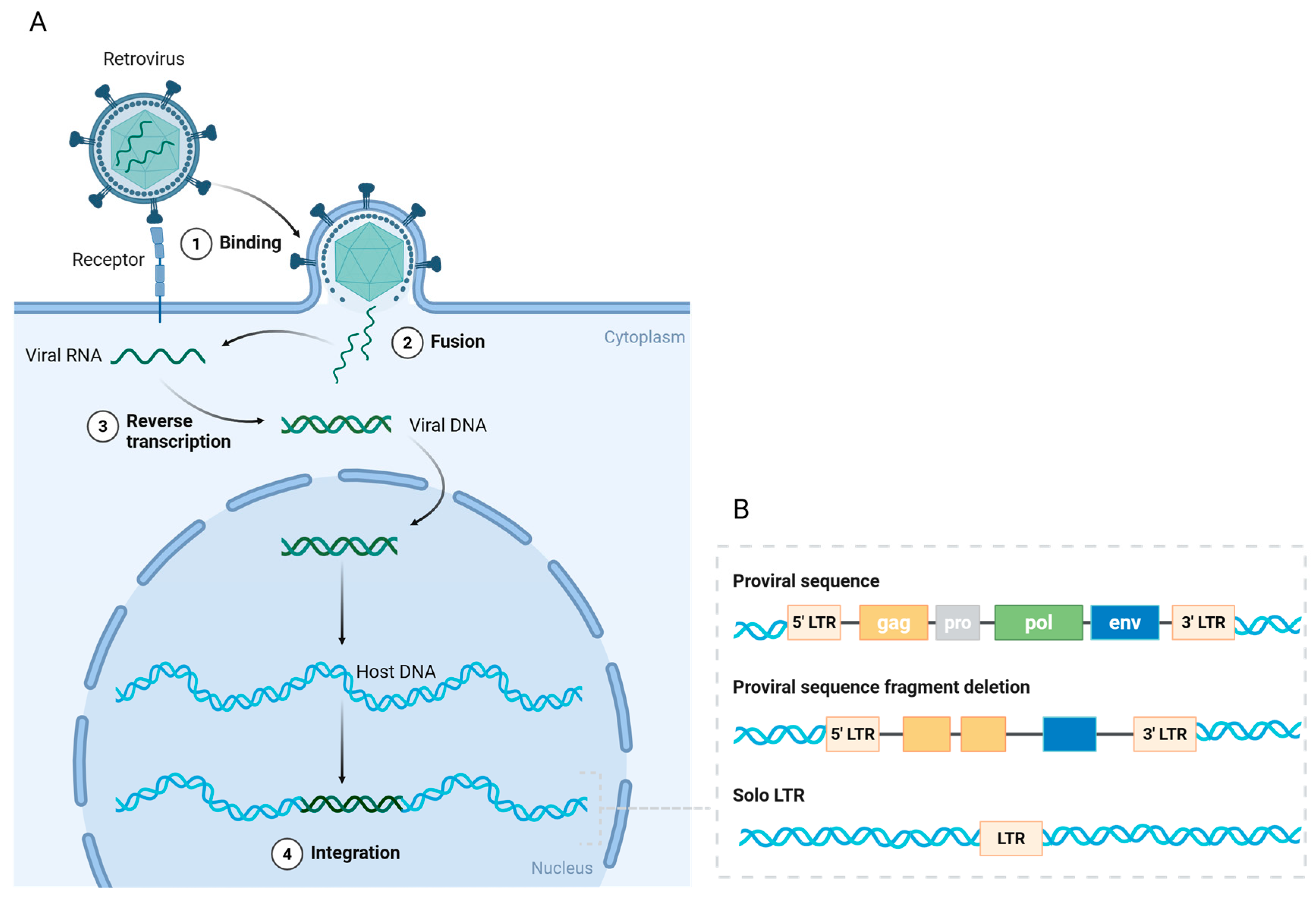
3. The Classification, Structure, and Function of HERVs
4. Human Endogenous Retrovirus K
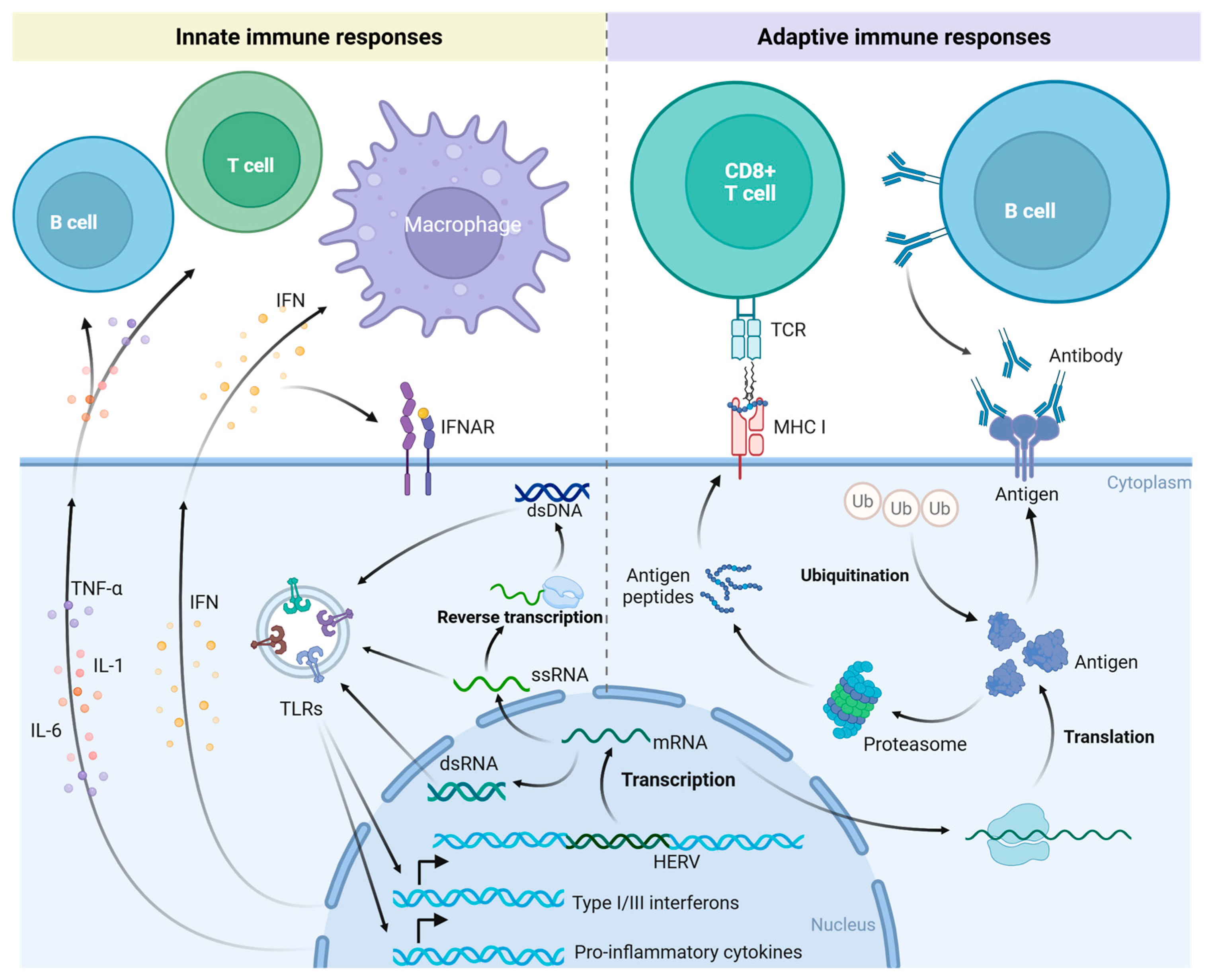
5. HERVs Trigger Immune Responses
6. The Association of HERV Expression and Melanoma
6.1. Activation of HERVs
6.2. Oncogenic Role of HERV-Derived Proteins
6.3. HERVs as a Biomarker of Melanoma
6.4. HERVs May Be Associated with the Stem Cell Phenotype of Melanoma Cells
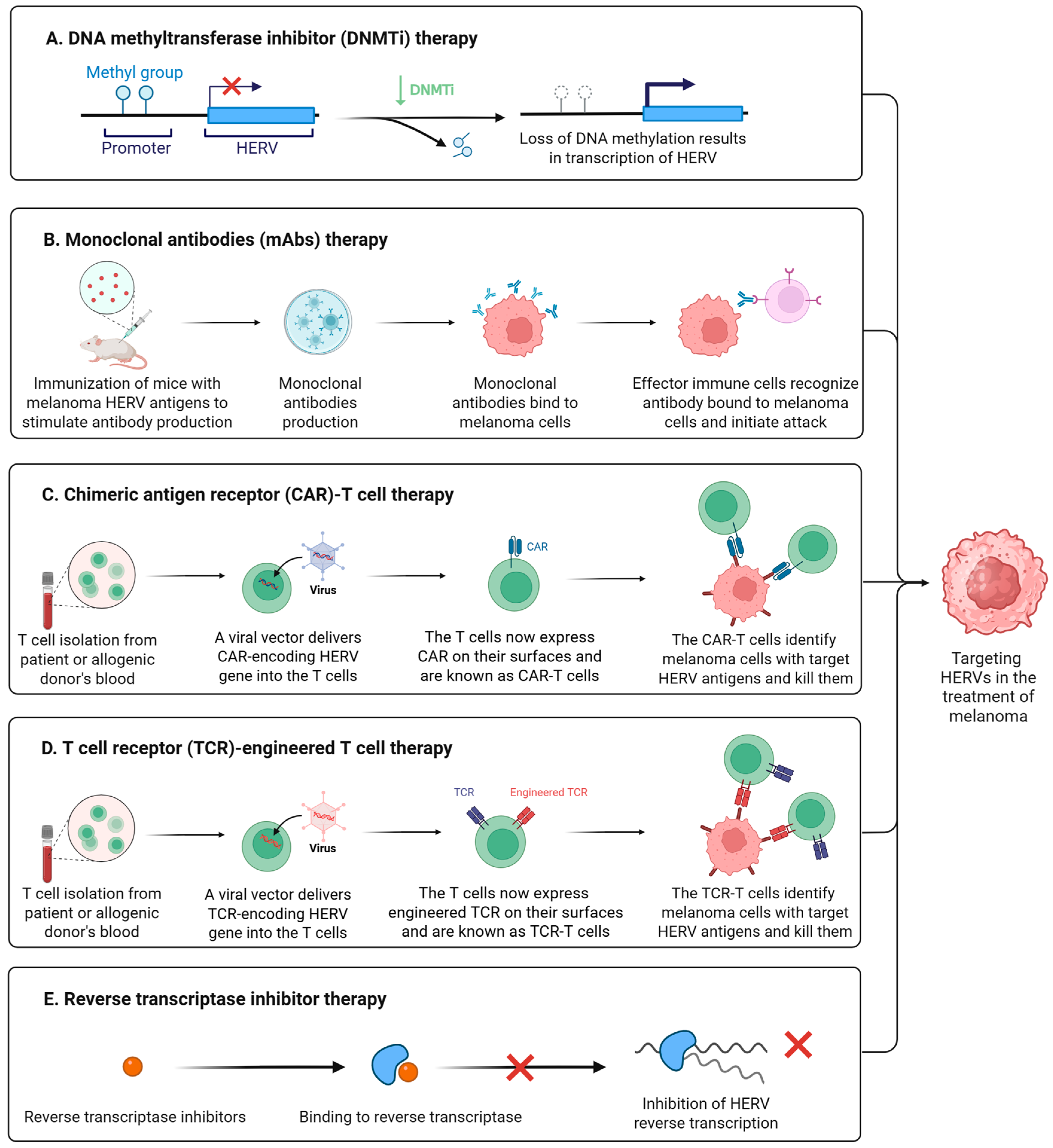
7. Therapeutic Implications of HERVs in Melanoma
7.1. Epigenetic Drugs Activate HERVs
7.2. Monoclonal Antibodies Targeting HERVs
7.3. CAR-T/TCR-T Therapy Targeting HERVs
7.4. Antiretroviral Drugs Inhibit HERV Activation
8. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous melanoma. Lancet 2023, 402, 485–502. [Google Scholar] [CrossRef]
- Dupin, E.; Le Douarin, N.M. Development of melanocyte precursors from the vertebrate neural crest. Oncogene 2003, 22, 3016–3023. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Testori, A.A.E.; Ribero, S.; Indini, A.; Mandalà, M. Adjuvant Treatment of Melanoma: Recent Developments and Future Perspectives. Am. J. Clin. Dermatol. 2019, 20, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, Reviews1017. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genom. Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Stoye, J.P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA 2004, 101 (Suppl. 2), 14572–14579. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Blaise, S.; de Parseval, N.; Bénit, L.; Heidmann, T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef]
- Wang, T.; Medynets, M.; Johnson, K.R.; Doucet-O’Hare, T.T.; DiSanza, B.; Li, W.; Xu, Y.; Bagnell, A.; Tyagi, R.; Sampson, K.; et al. Regulation of stem cell function and neuronal differentiation by HERV-K via mTOR pathway. Proc. Natl. Acad. Sci. USA 2020, 117, 17842–17853. [Google Scholar] [CrossRef]
- Dopkins, N.; Nixon, D.F. Activation of human endogenous retroviruses and its physiological consequences. Nat. Rev. Mol. Cell Biol. 2024, 25, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.C.; Marston, J.L.; Iñiguez, L.P.; Bendall, M.L.; Chiappinelli, K.B.; Nixon, D.F.; Crandall, K.A. Locus-Specific Characterization of Human Endogenous Retrovirus Expression in Prostate, Breast, and Colon Cancers. Cancer Res. 2021, 81, 3449–3460. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Holmes, D. The cancer that rises with the sun. Nature 2014, 515, S110–S111. [Google Scholar] [CrossRef]
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Brochez, L.; Del Marmol, V.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics—Update 2024. Eur. J. Cancer 2025, 215, 115152. [Google Scholar] [CrossRef] [PubMed]
- Dzwierzynski, W.W. Managing malignant melanoma. Plast. Reconstr. Surg. 2013, 132, 446e–460e. [Google Scholar] [CrossRef] [PubMed]
- Pavri, S.N.; Clune, J.; Ariyan, S.; Narayan, D. Malignant Melanoma: Beyond the Basics. Plast. Reconstr. Surg. 2016, 138, 330e–340e. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Brochez, L.; Del Marmol, V.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment—Update 2024. Eur. J. Cancer 2025, 215, 115153. [Google Scholar] [CrossRef]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Picconi, O.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur. J. Cancer 2005, 41, 45–60. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Stickley, M.; Watt, P.; Hughes, M.C.; Davis, M.B.; Green, A.C. Anatomic site, sun exposure, and risk of cutaneous melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3172–3177. [Google Scholar] [CrossRef]
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11, 4306. [Google Scholar] [CrossRef]
- Ribeiro Moura Brasil Arnaut, J.; Dos Santos Guimarães, I.; Evangelista Dos Santos, A.C.; de Moraes Lino da Silva, F.; Machado, J.R.; de Melo, A.C. Molecular landscape of Hereditary Melanoma. Crit. Rev. Oncol. /Hematol. 2021, 164, 103425. [Google Scholar] [CrossRef]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, H.; Zheng, S.; Han, R.; Wu, K.; Tang, S.; Zhong, X.; Chen, J. Elucidating tobacco smoke-induced craniofacial deformities: Biomarker and MAPK signaling dysregulation unraveled by cross-species multi-omics analysis. Ecotoxicol. Environ. Saf. 2024, 288, 117343. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Seth, R.; Agarwala, S.S.; Messersmith, H.; Alluri, K.C.; Ascierto, P.A.; Atkins, M.B.; Bollin, K.; Chacon, M.; Davis, N.; Faries, M.B.; et al. Systemic Therapy for Melanoma: ASCO Guideline Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 4794–4820. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Mc, C.B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Weiss, R.A. The discovery of endogenous retroviruses. Retrovirology 2006, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; Bryan, T.; Rasheed, S.; Khan, A.S. Identification and cloning of endogenous retroviral sequences present in human DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 4892–4896. [Google Scholar] [CrossRef]
- Leib-Mösch, C.; Brack-Werner, R.; Werner, T.; Bachmann, M.; Faff, O.; Erfle, V.; Hehlmann, R. Endogenous retroviral elements in human DNA. Cancer Res. 1990, 50 (17 Suppl), 5636s–5642s. [Google Scholar]
- Belshaw, R.; Pereira, V.; Katzourakis, A.; Talbot, G.; Paces, J.; Burt, A.; Tristem, M. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2004, 101, 4894–4899. [Google Scholar] [CrossRef]
- Nelson, P.N.; Carnegie, P.R.; Martin, J.; Davari Ejtehadi, H.; Hooley, P.; Roden, D.; Rowland-Jones, S.; Warren, P.; Astley, J.; Murray, P.G. Demystified. Human endogenous retroviruses. Mol. Pathol. 2003, 56, 11–18. [Google Scholar] [CrossRef]
- Mager, D.L.; Medstrand, P. Retroviral repeat sequences. Nat. Encycl. Hum. Genome 2003, 5, 57–63. [Google Scholar]
- Jakobsson, J.; Vincendeau, M. SnapShot: Human endogenous retroviruses. Cell 2022, 185, 400–400.e1. [Google Scholar] [CrossRef]
- Hughes, J.F.; Coffin, J.M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 1668–1672. [Google Scholar] [CrossRef]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef]
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.; Göke, J.; Bourque, G.; Ng, H.H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Medstrand, P.; Mager, D.L. An endogenous retroviral long terminal repeat is the dominant promoter for human beta1,3-galactosyltransferase 5 in the colon. Proc. Natl. Acad. Sci. USA 2003, 100, 12841–12846. [Google Scholar] [CrossRef]
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L.; Stoye, J.P. Mammalian Endogenous Retroviruses. Microbiol. Spectr. 2015, 3, Mdna3-0009-2014. [Google Scholar] [CrossRef]
- Stocking, C.; Kozak, C.A. Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef]
- Sexton, C.E.; Tillett, R.L.; Han, M.V. The essential but enigmatic regulatory role of HERVH in pluripotency. Trends Genet. 2022, 38, 12–21. [Google Scholar] [CrossRef]
- Brattås, P.L.; Jönsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Hantak, M.P.; Einstein, J.; Kearns, R.B.; Shepherd, J.D. Intercellular Communication in the Nervous System Goes Viral. Trends Neurosci. 2021, 44, 248–259. [Google Scholar] [CrossRef]
- Hanna, C.W.; Pérez-Palacios, R.; Gahurova, L.; Schubert, M.; Krueger, F.; Biggins, L.; Andrews, S.; Colomé-Tatché, M.; Bourc’his, D.; Dean, W.; et al. Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol. 2019, 20, 225. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, E.; Magiorkinis, G. Interplay between endogenous and exogenous human retroviruses. Trends Microbiol. 2023, 31, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Schanab, O.; Humer, J.; Gleiss, A.; Mikula, M.; Sturlan, S.; Grunt, S.; Okamoto, I.; Muster, T.; Pehamberger, H.; Waltenberger, A. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011, 24, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Zheng, M.; Yu, Y.; Wu, J.; Kuang, Q.; Hu, Z.; Ouyang, L.; Lu, S.; Zhao, M. Ultraviolet light induces HERV expression to activate RIG-I signalling pathway in keratinocytes. Exp. Dermatol. 2022, 31, 1165–1176. [Google Scholar] [CrossRef]
- Lima-Junior, D.S.; Krishnamurthy, S.R.; Bouladoux, N.; Collins, N.; Han, S.J.; Chen, E.Y.; Constantinides, M.G.; Link, V.M.; Lim, A.I.; Enamorado, M.; et al. Endogenous retroviruses promote homeostatic and inflammatory responses to the microbiota. Cell 2021, 184, 3794–3811.e19. [Google Scholar] [CrossRef]
- Jasemi, S.; Simula, E.R.; Pantaleo, A.; Sechi, L.A. Transcriptional Upregulation of HERV-env Genes Under Simulated Microgravity. Viruses 2025, 17, 306. [Google Scholar] [CrossRef]
- Kassiotis, G. The Immunological Conundrum of Endogenous Retroelements. Annu. Rev. Immunol. 2023, 41, 99–125. [Google Scholar] [CrossRef]
- Jansz, N.; Faulkner, G.J. Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 2021, 22, 147. [Google Scholar] [CrossRef]
- Ono, M.; Yasunaga, T.; Miyata, T.; Ushikubo, H. Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J. Virol. 1986, 60, 589–598. [Google Scholar] [CrossRef]
- Vargiu, L.; Rodriguez-Tomé, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 2016, 13, 7. [Google Scholar] [CrossRef]
- Moyes, D.; Griffiths, D.J.; Venables, P.J. Insertional polymorphisms: A new lease of life for endogenous retroviruses in human disease. Trends Genet. 2007, 23, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Belshaw, R.; Dawson, A.L.; Woolven-Allen, J.; Redding, J.; Burt, A.; Tristem, M. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): Implications for present-day activity. J. Virol. 2005, 79, 12507–12514. [Google Scholar] [CrossRef] [PubMed]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lin, L.; Malhotra, R.; Yang, L.; Acharya, R.; Poss, M. A computational framework to assess genome-wide distribution of polymorphic human endogenous retrovirus-K In human populations. PLoS Comput. Biol. 2019, 15, e1006564. [Google Scholar] [CrossRef]
- Schmitt, K.; Reichrath, J.; Roesch, A.; Meese, E.; Mayer, J. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol. Evol. 2013, 5, 307–328. [Google Scholar] [CrossRef]
- Turner, G.; Barbulescu, M.; Su, M.; Jensen-Seaman, M.I.; Kidd, K.K.; Lenz, J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 2001, 11, 1531–1535. [Google Scholar] [CrossRef]
- Burn, A.; Roy, F.; Freeman, M.; Coffin, J.M. Widespread expression of the ancient HERV-K (HML-2) provirus group in normal human tissues. PLoS Biol. 2022, 20, e3001826. [Google Scholar] [CrossRef]
- Schmitt, K.; Heyne, K.; Roemer, K.; Meese, E.; Mayer, J. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mob. DNA 2015, 6, 4. [Google Scholar] [CrossRef]
- Löwer, R.; Tönjes, R.R.; Korbmacher, C.; Kurth, R.; Löwer, J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J. Virol. 1995, 69, 141–149. [Google Scholar] [CrossRef]
- Armbruester, V.; Sauter, M.; Krautkraemer, E.; Meese, E.; Kleiman, A.; Best, B.; Roemer, K.; Mueller-Lantzsch, N. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1800–1807. [Google Scholar]
- Chen, T.; Meng, Z.; Gan, Y.; Wang, X.; Xu, F.; Gu, Y.; Xu, X.; Tang, J.; Zhou, H.; Zhang, X.; et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating β-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia 2013, 27, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Hanke, K.; Chudak, C.; Kurth, R.; Bannert, N. The Rec protein of HERV-K(HML-2) upregulates androgen receptor activity by binding to the human small glutamine-rich tetratricopeptide repeat protein (hSGT). Int. J. Cancer 2013, 132, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Denne, M.; Sauter, M.; Armbruester, V.; Licht, J.D.; Roemer, K.; Mueller-Lantzsch, N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J. Virol. 2007, 81, 5607–5616. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, C.; Tsang, J.; Bireau, C.; Heidmann, T.; Dewannieux, M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017, 13, e1006451. [Google Scholar] [CrossRef]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef]
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Grønbæk, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2183–2191. [Google Scholar] [CrossRef]
- Seifarth, W.; Frank, O.; Zeilfelder, U.; Spiess, B.; Greenwood, A.D.; Hehlmann, R.; Leib-Mösch, C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 2005, 79, 341–352. [Google Scholar] [CrossRef]
- Rauch, E.; Amendt, T.; Lopez Krol, A.; Lang, F.B.; Linse, V.; Hohmann, M.; Keim, A.C.; Kreutzer, S.; Kawengian, K.; Buchholz, M.; et al. T-bet(+) B cells are activated by and control endogenous retroviruses through TLR-dependent mechanisms. Nat. Commun. 2024, 15, 1229. [Google Scholar] [CrossRef]
- Young, G.R.; Eksmond, U.; Salcedo, R.; Alexopoulou, L.; Stoye, J.P.; Kassiotis, G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature 2012, 491, 774–778. [Google Scholar] [CrossRef]
- Yu, P.; Lübben, W.; Slomka, H.; Gebler, J.; Konert, M.; Cai, C.; Neubrandt, L.; Prazeres da Costa, O.; Paul, S.; Dehnert, S.; et al. Nucleic acid-sensing Toll-like receptors are essential for the control of endogenous retrovirus viremia and ERV-induced tumors. Immunity 2012, 37, 867–879. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725. [Google Scholar] [CrossRef]
- Arru, G.; Galleri, G.; Deiana, G.A.; Zarbo, I.R.; Sechi, E.; Bo, M.; Cadoni, M.P.L.; Corda, D.G.; Frau, C.; Simula, E.R.; et al. HERV-K Modulates the Immune Response in ALS Patients. Microorganisms 2021, 9, 1784. [Google Scholar] [CrossRef]
- Dopkins, N.; O’Mara, M.M.; Lawrence, E.; Fei, T.; Sandoval-Motta, S.; Nixon, D.F.; Bendall, M.L. A field guide to endogenous retrovirus regulatory networks. Mol. Cell 2022, 82, 3763–3768. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Radvanyi, L.; Rycaj, K.; Plummer, J.B.; Yan, P.; Sastry, K.J.; Piyathilake, C.J.; Hunt, K.K.; Johanning, G.L. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008, 68, 5869–5877. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Hsu, C.; Mondesire, W.; Parker, L.L.; Wang, G.; Overwijk, W.W.; Lapointe, R.; Yang, J.C.; Wang, R.F.; Restifo, N.P.; et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001, 61, 7920–7924. [Google Scholar]
- Büscher, K.; Trefzer, U.; Hofmann, M.; Sterry, W.; Kurth, R.; Denner, J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65, 4172–4180. [Google Scholar] [CrossRef]
- Humer, J.; Waltenberger, A.; Grassauer, A.; Kurz, M.; Valencak, J.; Rapberger, R.; Hahn, S.; Löwer, R.; Wolff, K.; Bergmann, M.; et al. Identification of a melanoma marker derived from melanoma-associated endogenous retroviruses. Cancer Res. 2006, 66, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Wang-Johanning, F.; Liu, J.; Rycaj, K.; Huang, M.; Tsai, K.; Rosen, D.G.; Chen, D.T.; Lu, D.W.; Barnhart, K.F.; Johanning, G.L. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 2007, 120, 81–90. [Google Scholar] [CrossRef]
- Arru, G.; Mameli, G.; Deiana, G.A.; Rassu, A.L.; Piredda, R.; Sechi, E.; Caggiu, E.; Bo, M.; Nako, E.; Urso, D.; et al. Humoral immunity response to human endogenous retroviruses K/W differentiates between amyotrophic lateral sclerosis and other neurological diseases. Eur. J. Neurol. 2018, 25, 1076-e84. [Google Scholar] [CrossRef]
- Nelson, P.N.; Roden, D.; Nevill, A.; Freimanis, G.L.; Trela, M.; Ejtehadi, H.D.; Bowman, S.; Axford, J.; Veitch, A.M.; Tugnet, N.; et al. Rheumatoid arthritis is associated with IgG antibodies to human endogenous retrovirus gag matrix: A potential pathogenic mechanism of disease? J. Rheumatol. 2014, 41, 1952–1960. [Google Scholar] [CrossRef]
- Simula, E.R.; Zarbo, I.R.; Arru, G.; Sechi, E.; Meloni, R.; Deiana, G.A.; Solla, P.; Sechi, L.A. Antibody Response to HERV-K and HERV-W Envelope Epitopes in Patients with Myasthenia Gravis. Int. J. Mol. Sci. 2023, 25, 446. [Google Scholar] [CrossRef] [PubMed]
- Ruberto, S.; Domınguez-Mozo, M.I.; Garcıa-Martınez, M.A.; Cossu, D.; Sechi, L.A.; Alvarez-Lafuente, R. Immune response profiling of HERV-W envelope proteins in multiple sclerosis: Potential biomarkers for disease progression. Front. Immunol. 2024, 15, 1505239. [Google Scholar] [CrossRef] [PubMed]
- Chisca, M.; Larouche, J.D.; Xing, Q.; Kassiotis, G. Antibodies against endogenous retroviruses. Immunol. Rev. 2024, 328, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Wang-Johanning, F.; Rycaj, K.; Plummer, J.B.; Li, M.; Yin, B.; Frerich, K.; Garza, J.G.; Shen, J.; Lin, K.; Yan, P.; et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J. Natl. Cancer Inst. 2012, 104, 189–210. [Google Scholar] [CrossRef]
- Ng, K.W.; Boumelha, J.; Enfield, K.S.S.; Almagro, J.; Cha, H.; Pich, O.; Karasaki, T.; Moore, D.A.; Salgado, R.; Sivakumar, M.; et al. Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature 2023, 616, 563–573. [Google Scholar] [CrossRef]
- Reis, B.S.; Jungbluth, A.A.; Frosina, D.; Holz, M.; Ritter, E.; Nakayama, E.; Ishida, T.; Obata, Y.; Carver, B.; Scher, H.; et al. Prostate cancer progression correlates with increased humoral immune response to a human endogenous retrovirus GAG protein. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6112–6125. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Li, M.; Esteva, F.J.; Hess, K.R.; Yin, B.; Rycaj, K.; Plummer, J.B.; Garza, J.G.; Ambs, S.; Johanning, G.L. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int. J. Cancer 2014, 134, 587–595. [Google Scholar] [CrossRef]
- Kleiman, A.; Senyuta, N.; Tryakin, A.; Sauter, M.; Karseladze, A.; Tjulandin, S.; Gurtsevitch, V.; Mueller-Lantzsch, N. HERV-K(HML-2) GAG/ENV antibodies as indicator for therapy effect in patients with germ cell tumors. Int. J. Cancer 2004, 110, 459–461. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Yura, M.; Yamamoto, R.; Kawakami, Y. Induction of immunoregulatory CD271+ cells by metastatic tumor cells that express human endogenous retrovirus H. Cancer Res. 2014, 74, 1361–1370. [Google Scholar] [CrossRef]
- Li, M.; Radvanyi, L.; Yin, B.; Rycaj, K.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zhang, Q.; Wang, Y.; Zhuang, Y.; Xu, L.; Ma, X.; Guan, D.; Zhou, J.; Liu, J.; Wu, X.; et al. TERT activates endogenous retroviruses to promote an immunosuppressive tumour microenvironment. EMBO Rep. 2022, 23, e52984. [Google Scholar] [CrossRef] [PubMed]
- Birkmayer, G.D.; Balda, B.R.; Miller, F.; Braun-Falco, O. Virus-like particles in metastases of human malignant melanoma. Die Naturwissenschaften 1972, 59, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Balda, B.R.; Birkmayer, G.D. Further evidence for viral etiology of human melanoma. Die Naturwissenschaften 1973, 60, 304. [Google Scholar] [CrossRef]
- Parsons, P.G.; Goss, P.; Pope, J.H. Detection in human melanoma cell lines of particles with some properties in common with RNA tumour viruses. Int. J. Cancer 1974, 13, 606–618. [Google Scholar] [CrossRef]
- Balda, B.R.; Hehlmann, R.; Cho, J.R.; Spiegelman, S. Oncornavirus-like particles in human skin cancers. Proc. Natl. Acad. Sci. USA 1975, 72, 3697–3700. [Google Scholar] [CrossRef]
- Schiavetti, F.; Thonnard, J.; Colau, D.; Boon, T.; Coulie, P.G. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002, 62, 5510–5516. [Google Scholar]
- Muster, T.; Waltenberger, A.; Grassauer, A.; Hirschl, S.; Caucig, P.; Romirer, I.; Födinger, D.; Seppele, H.; Schanab, O.; Magin-Lachmann, C.; et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003, 63, 8735–8741. [Google Scholar]
- Sciamanna, I.; Landriscina, M.; Pittoggi, C.; Quirino, M.; Mearelli, C.; Beraldi, R.; Mattei, E.; Serafino, A.; Cassano, A.; Sinibaldi-Vallebona, P.; et al. Inhibition of endogenous reverse transcriptase antagonizes human tumor growth. Oncogene 2005, 24, 3923–3931. [Google Scholar] [CrossRef]
- Büscher, K.; Hahn, S.; Hofmann, M.; Trefzer, U.; Ozel, M.; Sterry, W.; Löwer, J.; Löwer, R.; Kurth, R.; Denner, J. Expression of the human endogenous retrovirus-K transmembrane envelope, Rec and Np9 proteins in melanomas and melanoma cell lines. Melanoma Res. 2006, 16, 223–234. [Google Scholar] [CrossRef]
- Lavie, L.; Kitova, M.; Maldener, E.; Meese, E.; Mayer, J. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J. Virol. 2005, 79, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Stengel, S.; Fiebig, U.; Kurth, R.; Denner, J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer 2010, 49, 401–411. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Z.; Wu, Z.; Ren, J.; Fan, Y.; Sun, L.; Cao, G.; Niu, Y.; Zhang, B.; Ji, Q.; et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell 2023, 186, 287–304.e26. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed]
- Katoh, I.; Mírová, A.; Kurata, S.; Murakami, Y.; Horikawa, K.; Nakakuki, N.; Sakai, T.; Hashimoto, K.; Maruyama, A.; Yonaga, T.; et al. Activation of the long terminal repeat of human endogenous retrovirus K by melanoma-specific transcription factor MITF-M. Neoplasia 2011, 13, 1081–1092. [Google Scholar] [CrossRef]
- Green, C.D.; Huang, Y.; Dou, X.; Yang, L.; Liu, Y.; Han, J.J. Impact of Dietary Interventions on Noncoding RNA Networks and mRNAs Encoding Chromatin-Related Factors. Cell Rep. 2017, 18, 2957–2968. [Google Scholar] [CrossRef]
- Lee, J.R.; Ahn, K.; Kim, Y.J.; Jung, Y.D.; Kim, H.S. Radiation-induced human endogenous retrovirus (HERV)-R env gene expression by epigenetic control. Radiat. Res. 2012, 178, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Cai, H.; Bunse, M.; Feschotte, C.; Izsvák, Z. Human Endogenous Retrovirus K Rec forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses 2020, 12, 1303. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Serafino, A.; Balestrieri, E.; Pierimarchi, P.; Matteucci, C.; Moroni, G.; Oricchio, E.; Rasi, G.; Mastino, A.; Spadafora, C.; Garaci, E.; et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell Res. 2009, 315, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Oricchio, E.; Sciamanna, I.; Beraldi, R.; Tolstonog, G.V.; Schumann, G.G.; Spadafora, C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene 2007, 26, 4226–4233. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Li, Z.; Wan, X.; Wang, Y.; Dong, J. Human endogenous retroviral K element encodes fusogenic activity in melanoma cells. J. Carcinog. 2013, 12, 5. [Google Scholar] [PubMed]
- Ying, H.; Xu, J.; Zhang, X.; Liang, T.; Bai, X. Human endogenous retrovirus-H long terminal repeat-associating 2: The next immune checkpoint for antitumour therapy. EBioMedicine 2022, 79, 103987. [Google Scholar] [CrossRef]
- Shattuck-Brandt, R.L.; Richmond, A. Enhanced degradation of I-kappaB alpha contributes to endogenous activation of NF-kappaB in Hs294T melanoma cells. Cancer Res. 1997, 57, 3032–3039. [Google Scholar]
- Zhao, R.; Chinai, J.M.; Buhl, S.; Scandiuzzi, L.; Ray, A.; Jeon, H.; Ohaegbulam, K.C.; Ghosh, K.; Zhao, A.; Scharff, M.D.; et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc. Natl. Acad. Sci. USA 2013, 110, 9879–9884. [Google Scholar] [CrossRef]
- Janakiram, M.; Chinai, J.M.; Fineberg, S.; Fiser, A.; Montagna, C.; Medavarapu, R.; Castano, E.; Jeon, H.; Ohaegbulam, K.C.; Zhao, R.; et al. Expression, Clinical Significance, and Receptor Identification of the Newest B7 Family Member HHLA2 Protein. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2359–2366. [Google Scholar] [CrossRef]
- Bhatt, R.S.; Berjis, A.; Konge, J.C.; Mahoney, K.M.; Klee, A.N.; Freeman, S.S.; Chen, C.H.; Jegede, O.A.; Catalano, P.J.; Pignon, J.C.; et al. KIR3DL3 Is an Inhibitory Receptor for HHLA2 that Mediates an Alternative Immunoinhibitory Pathway to PD1. Cancer Immunol. Res. 2021, 9, 156–169. [Google Scholar] [CrossRef]
- Huang, F.X.; Wu, J.W.; Cheng, X.Q.; Wang, J.H.; Wen, X.Z.; Li, J.J.; Zhang, Q.; Jiang, H.; Ding, Q.Y.; Zhu, X.F.; et al. HHLA2 predicts improved prognosis of anti-PD-1/PD-L1 immunotherapy in patients with melanoma. Front. Immunol. 2022, 13, 902167. [Google Scholar] [CrossRef]
- Hahn, S.; Ugurel, S.; Hanschmann, K.M.; Strobel, H.; Tondera, C.; Schadendorf, D.; Löwer, J.; Löwer, R. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res. Hum. Retroviruses 2008, 24, 717–723. [Google Scholar] [CrossRef]
- Bendall, M.L.; Francis, J.H.; Shoushtari, A.N.; Nixon, D.F. Specific human endogenous retroviruses predict metastatic potential in uveal melanoma. JCI Insight 2022, 7, e147172. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhou, L.; Jiang, J.; Chen, J.; Geng, D.; Chen, Y.; Han, X.; Xie, Q.; Guo, G.; Chen, X.; et al. Induction of the p21/CDK6 pathway and alteration of the immune microenvironment by the stem cell marker CBX3 in melanoma. Stem Cell Res. Ther. 2025, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Argaw-Denboba, A.; Balestrieri, E.; Serafino, A.; Cipriani, C.; Bucci, I.; Sorrentino, R.; Sciamanna, I.; Gambacurta, A.; Sinibaldi-Vallebona, P.; Matteucci, C. HERV-K activation is strictly required to sustain CD133+ melanoma cells with stemness features. J. Exp. Clin. Cancer Res. 2017, 36, 20. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, A.; Balestrieri, E.; Petrone, V.; Argaw-Denboba, A.; Cipriani, C.; Miele, M.T.; Grelli, S.; Sinibaldi-Vallebona, P.; Matteucci, C. The Concomitant Expression of Human Endogenous Retroviruses and Embryonic Genes in Cancer Cells under Microenvironmental Changes is a Potential Target for Antiretroviral Drugs. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2019, 12, 105–118. [Google Scholar] [CrossRef]
- Shah, A.H.; Rivas, S.R.; Doucet-O’Hare, T.T.; Govindarajan, V.; DeMarino, C.; Wang, T.; Ampie, L.; Zhang, Y.; Banasavadi-Siddegowda, Y.K.; Walbridge, S.; et al. Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J. Clin. Investig. 2023, 133, e167929. [Google Scholar] [CrossRef]
- Hosseiniporgham, S.; Sechi, L.A. Anti-HERV-K Drugs and Vaccines, Possible Therapies against Tumors. Vaccines 2023, 11, 751. [Google Scholar] [CrossRef]
- Wolff, F.; Leisch, M.; Greil, R.; Risch, A.; Pleyer, L. The double-edged sword of (re)expression of genes by hypomethylating agents: From viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun. Signal. 2017, 15, 13. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, F. Therapeutic monoclonal antibodies. Lancet 2000, 355, 735–740. [Google Scholar] [CrossRef]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Rabinovich, B.A.; Mi, T.; Switzer, K.C.; Olivares, S.; Maiti, S.N.; Plummer, J.B.; Singh, H.; Kumaresan, P.R.; Huls, H.M.; et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Krishnamurthy, J.; Wei, Y.; Li, M.; Hunt, K.; Johanning, G.L.; Cooper, L.J.; Wang-Johanning, F. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology 2015, 4, e1047582. [Google Scholar] [CrossRef]
- Baulu, E.; Gardet, C.; Chuvin, N.; Depil, S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci. Adv. 2023, 9, eadf3700. [Google Scholar] [CrossRef]
- Bonaventura, P.; Alcazer, V.; Mutez, V.; Tonon, L.; Martin, J.; Chuvin, N.; Michel, E.; Boulos, R.E.; Estornes, Y.; Valladeau-Guilemond, J.; et al. Identification of shared tumor epitopes from endogenous retroviruses inducing high-avidity cytotoxic T cells for cancer immunotherapy. Sci. Adv. 2022, 8, eabj3671. [Google Scholar] [CrossRef] [PubMed]
- Denner, J. Endogenous retroviruses expressed in human tumours cannot be used as targets for anti-tumour vaccines. Transl. Oncol. 2021, 14, 100941. [Google Scholar] [CrossRef]
- Denner, J. Comment on: Endogenous retroviruses expressed in human tumours cannot be used as targets for anti-tumour vaccines. Transl. Oncol. 2021, 14, 101041. [Google Scholar] [CrossRef]
- Sinibaldi-Vallebona, P.; Lavia, P.; Garaci, E.; Spadafora, C. A role for endogenous reverse transcriptase in tumorigenesis and as a target in differentiating cancer therapy. Genes Chromosomes Cancer 2006, 45, 1–10. [Google Scholar] [CrossRef]
- Zanrè, V.; Bellinato, F.; Cardile, A.; Passarini, C.; Monticelli, J.; Di Bella, S.; Menegazzi, M. Lamivudine, Doravirine, and Cabotegravir Downregulate the Expression of Human Endogenous Retroviruses (HERVs), Inhibit Cell Growth, and Reduce Invasive Capability in Melanoma Cell Lines. Int. J. Mol. Sci. 2024, 25, 1615. [Google Scholar] [CrossRef] [PubMed]
- Zanrè, V.; Bellinato, F.; Cardile, A.; Passarini, C.; Di Bella, S.; Menegazzi, M. BRAF-Mutated Melanoma Cell Lines Develop Distinct Molecular Signatures After Prolonged Exposure to AZ628 or Dabrafenib: Potential Benefits of the Antiretroviral Treatments Cabotegravir or Doravirine on BRAF-Inhibitor-Resistant Cells. Int. J. Mol. Sci. 2024, 25, 11939. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Satta, R.R.; Simula, E.R.; Tang, S.; Molicotti, P.; Cossu, A.; Rubino, C.; Sechi, L.A. The Role of Human Endogenous Retroviruses in the Initiation and Progression of Melanoma. Biomedicines 2025, 13, 1662. https://doi.org/10.3390/biomedicines13071662
Lin Y, Satta RR, Simula ER, Tang S, Molicotti P, Cossu A, Rubino C, Sechi LA. The Role of Human Endogenous Retroviruses in the Initiation and Progression of Melanoma. Biomedicines. 2025; 13(7):1662. https://doi.org/10.3390/biomedicines13071662
Chicago/Turabian StyleLin, Yao, Rosanna Rita Satta, Elena Rita Simula, Shijie Tang, Paola Molicotti, Antonio Cossu, Corrado Rubino, and Leonardo Antonio Sechi. 2025. "The Role of Human Endogenous Retroviruses in the Initiation and Progression of Melanoma" Biomedicines 13, no. 7: 1662. https://doi.org/10.3390/biomedicines13071662
APA StyleLin, Y., Satta, R. R., Simula, E. R., Tang, S., Molicotti, P., Cossu, A., Rubino, C., & Sechi, L. A. (2025). The Role of Human Endogenous Retroviruses in the Initiation and Progression of Melanoma. Biomedicines, 13(7), 1662. https://doi.org/10.3390/biomedicines13071662