

Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy

Abstract
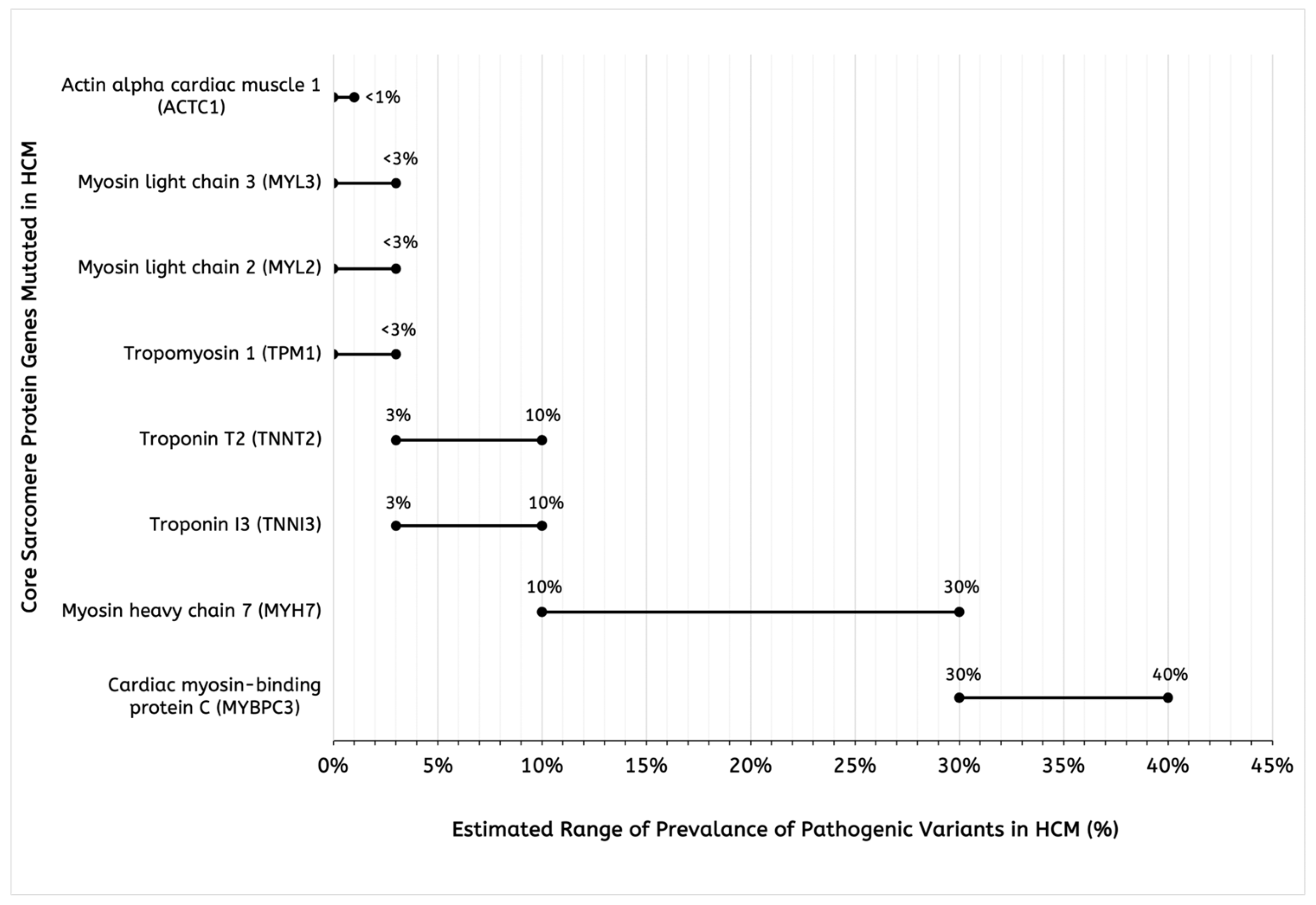
1. Introduction
2. Mechanism of Action
3. Clinical Efficacy
3.1. Mavacamten Clinical Trials
3.1.1. Phase II Trials: PIONEER-HCM
3.1.2. Phase II Trials: MAVERICK-HCM
3.1.3. Phase III Trials: EXPLORER-HCM
3.2. Aficamten Clinical Trials
3.2.1. Phase III Trials: SEQUOIA-HCM
3.2.2. Phase II Trials: REDWOOD-HCM
4. Ongoing Trials
5. Safety Profile
6. Clinical Insights
7. Future Directions
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HCM | hypertrophic cardiomyopathy |
| CMI | cardiac myosin inhibitors |
| LVOT | left ventricular outflow tract |
| oHCM | obstructive hypertrophic cardiomyopathy |
| nHCM | non-obstructive hypertrophic cardiomyopathy |
| ATP | adenosine triphosphate |
| LVEF | left ventricular ejection fraction |
| NYHA | New York Heart Association |
| ECG | electrocardiography |
| KCCQ-CSS | Kansas City Cardiomyopathy Questionnaire Clinical Summary Score |
| HCMSQ-SoB | Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness-of-Breath sub-score |
| HFpEF | heart failure with preserved ejection fraction |
| FDA | Food and Drug Administration |
| EMA | European Medicines Agency |
References
- Basit, H.; Alahmadi, M.H.; Rout, P.; Sharma, S. Hypertrophic Cardiomyopathy; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Zou, Y.; Song, L.; Wang, Z.; Ma, A.; Liu, T.; Gu, H.; Lu, S.; Wu, P.; Zhang, Y.; Shen, L.; et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: A population-based echocardiographic analysis of 8080 adults. Am. J. Med. 2004, 116, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Hada, Y.; Sakamoto, T.; Amano, K.; Yamaguchi, T.; Takenaka, K.; Takahashi, H.; Takikawa, R.; Hasegawa, I.; Takahashi, T.; Suzuki, J.-I.; et al. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am. J. Cardiol. 1987, 59, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Mathenge, R.; Casey, S.A.; Poliac, L.C.; Longe, T.F. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities11Each of the authors contributed significantly to the submitted work including: (1) conception and design of the project and/or interpretation of data; (2) drafting and/or revising the manuscript, and (3) final approval of the submitted manuscript. J. Am. Coll. Cardiol. 1999, 33, 1590–1595. [Google Scholar] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Jensen, M.K.; Havndrup, O.; Christiansen, M.; Andersen, P.S.; Diness, B.; Axelsson, A.; Bundgaard, H. Penetrance of Hypertrophic Cardiomyopathy in Children and Adolescents. Circulation 2013, 127, 48–54. [Google Scholar] [CrossRef]
- Frey, N.; Luedde, M.; Katus, H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 91–100. [Google Scholar] [CrossRef]
- Ho, C.Y. Genetics and Clinical Destiny: Improving Care in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2430–2440. [Google Scholar] [CrossRef]
- Borrelli, F.; Losi, M.A.; Canciello, G.; Todde, G.; Perillo, E.F.; Ordine, L.; Frisso, G.; Esposito, G.; Lombardi, R. Sarcomeric versus Non-Sarcomeric HCM. Cardiogenetics 2023, 13, 92–105. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar]
- Topriceanu, C.C.; Pereira, A.C.; Moon, J.C.; Captur, G.; Ho, C.Y. Meta-Analysis of Penetrance and Systematic Review on Transition to Disease in Genetic Hypertrophic Cardiomyopathy. Circulation 2024, 149, 107–123. [Google Scholar] [CrossRef]
- Guo, L.; Ma, Z.; Yang, W.; Zhang, F.; Shao, H.; Liu, L.; Gao, C.; Tao, L. Identifying Obstructive Hypertrophic Cardiomyopathy from Nonobstructive Hypertrophic Cardiomyopathy: Development and Validation of a Model Based on Electrocardiogram Features. Glob. Heart 2023, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Pozios, I.; Haileselassie, B.; Ventoulis, I.; Liu, H.; Sorensen, L.L.; Canepa, M.; Phillip, S.; Abraham, M.R.; Abraham, T.P. Clinical Outcomes in Patients with Nonobstructive, Labile, and Obstructive Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e006657. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef]
- Flamm, M.D.; Harrison, D.C.; Hancock, E.W. Muscular Subaortic Stenosis. Circulation 1968, 38, 846–858. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Iavarone, M.; Monda, E.; Vritz, O.; Albert, D.C.; Rubino, M.; Verrillo, F.; Caiazza, M.; Lioncino, M.; Amodio, F.; Guarnaccia, N.; et al. Medical treatment of patients with hypertrophic cardiomyopathy: An overview of current and emerging therapy. Arch. Cardiovasc. Dis. 2022, 115, 529–537. [Google Scholar] [CrossRef]
- Schaff, H.V.; Juarez-Casso, F.M. Treatment Strategies for Hypertrophic Cardiomyopathy: Surgical. Am. J. Cardiol. 2024, 212, S53–S63. [Google Scholar] [CrossRef]
- Haddy, S. Anesthesia for Structural Heart Interventions. Cardiol. Clin. 2013, 31, 455–465. [Google Scholar] [CrossRef]
- Veselka, J.; Duchonová, R.; Procházková, S.; Homolová, I.; Pálenícková, J.; Zemánek, D.; Pernisová, Z.; Tesar, D. The biphasic course of changes of left ventricular outflow gradient after alcohol septal ablation for hypertrophic obstructive cardiomyopathy. Kardiol. Pol. 2004, 60, 133–136; discussion 137. [Google Scholar]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Lorenzini, M.; Anastasiou, Z.; O’mAhony, C.; Guttman, O.P.; Gimeno, J.R.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Garcia-Pavia, P.; et al. Mortality Among Referral Patients with Hypertrophic Cardiomyopathy vs the General European Population. JAMA Cardiol. 2020, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.J.; Hwee, D.T.; Robert-Paganin, J.; Chuang, C.; Chin, E.R.; Edell, S.; Lee, K.H.; Madhvani, R.; Paliwal, P.; Pernier, J.; et al. Aficamten is a small-molecule cardiac myosin inhibitor designed to treat hypertrophic cardiomyopathy. Nat. Cardiovasc. Res. 2024, 3, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Bello, J.; Pellegrini, M.V. Mavacamten; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Masri, A.; Olivotto, I. Cardiac Myosin Inhibitors as a Novel Treatment Option for Obstructive Hypertrophic Cardiomyopathy: Addressing the Core of the Matter. J. Am. Heart Assoc. 2022, 11, e024656. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Ramadan, M.M.; Al-Najjar, R.A.; Abady, R.S.; Obaid, H.A.; Mostafa, Y.A.; Al-Obeid, M.T.; Elmahal, M. Mavacamten Cardiac Myosin Inhibitor: Clinical Applications and Future Perspectives. Cureus 2025, 17, e82722. [Google Scholar] [CrossRef]
- McMillan, S.N.; Pitts, J.R.T.; Barua, B.; Winkelmann, D.A.; Scarff, C.A. Mavacamten inhibits myosin activity by stabilising the myosin interacting-heads motif and stalling motor force generation. bioRxiv 2025. [Google Scholar]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients with Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; Düngen, H.-D.; et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2024, 390, 1849–1861. [Google Scholar] [CrossRef]
- Masri, A.; Sherrid, M.V.; Abraham, T.P.; Choudhury, L.; Garcia-Pavia, P.; Kramer, C.M.; Barriales-Villa, R.; Owens, A.T.; Rader, F.; Nagueh, S.F.; et al. Efficacy and Safety of Aficamten in Symptomatic Nonobstructive Hypertrophic Cardiomyopathy: Results From the REDWOOD-HCM Trial, Cohort 4. J. Card. Fail. 2024, 30, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Nissen, S.E.; Abraham, T.; Olivotto, I.; Garcia-Pavia, P.; Lopes, R.D.; Verheyen, N.; Wever-Pinzon, O.; Wolski, K.; Jaber, W.; et al. Mavacamten in Symptomatic Nonobstructive Hypertrophic Cardiomyopathy. JACC Heart Fail. 2025, 13, 358–370. [Google Scholar] [CrossRef]
- BusinessWire. Bristol Myers Squibb Provides Update on Phase 3 ODYSSEY-HCM Trial; BusinessWire: San Francisco, CA, USA, 2025. [Google Scholar]
- Garcia-Pavia, P.; Bilen, O.; Burroughs, M.; Costabel, J.P.; Correia, E.d.B.; Dybro, A.M.; Elliott, P.; Lakdawala, N.K.; Mann, A.; Nair, A.; et al. Aficamten vs Metoprolol for Obstructive Hypertrophic Cardiomyopathy. JACC Heart Fail. 2025, 13, 346–357. [Google Scholar] [CrossRef]
- Weiser, D. Cytokinetics Announces Positive Topline Results From MAPLE-HCM; Cytokinetics: San Francisco, CA, USA, 2025. [Google Scholar]
- Cytokinetics, M.D. Phase 3 Trial to Evaluate the Efficacy and Safety of Aficamten Compared to Placebo in Adults with Symptomatic nHCM (ACACIA-HCM); Cytokinetics: San Francisco, CA, USA, 2025. [Google Scholar]
- Saberi, S.; Abraham, T.P.; Choudhury, L.; Owens, A.T.; Tower-Rader, A.; Rader, F.; Pavia, P.G.; Olivotto, I.; Coats, C.; Fifer, M.A.; et al. Long-Term Efficacy and Safety of Aficamten in Patients with Symptomatic Obstructive Hypertrophic Cardiomyopathy. JACC 2023, 81, 324. [Google Scholar] [CrossRef]
- Cytokinetics. Cytokinetics Announces Start of CEDAR-HCM, a Clinical Trial of Aficamten in a Pediatric Population with Symptomatic Obstructive Hypertrophic Cardiomyopathy; Cytokinetics: San Francisco, CA, USA, 2025. [Google Scholar]
- Davis, B.J.; Volk, H.; Nguyen, O.; Kamna, D.; Chen, H.; Barriales-Villa, R.; Garcia-Pavia, P.; Olivotto, I.; Owens, A.T.; Coats, C.J.; et al. Safety and Efficacy of Mavacamten and Aficamten in Patients with Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2025, 14, e038758. [Google Scholar] [CrossRef]
- European Medicines Agency. Camzyos Mavacamten; European Medicines Agency: Amsterdam, The Netherlands, 2025. [Google Scholar]
- Haraf, R.; Habib, H.; Masri, A. The Revolution of Cardiac Myosin Inhibitors in Patients with Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2024, 40, 800–819. [Google Scholar] [CrossRef]
- 214998Orig1s000; Center for Drug Evaluation and Research. FDA: Silver Spring, MD, USA, 2022.
- Coats, C.J.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Düngen, H.; Garcia-Pavia, P.; et al. Dosing and Safety Profile of Aficamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Results From SEQUOIA-HCM. J. Am. Heart Assoc. 2024, 13, e035993. [Google Scholar] [CrossRef]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Wal, M.V.; Wang, X.; Hwee, D.T.; Wu, Y.; Wang, J.; Chin, E.R.; et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef]
- Malik, F.I.; Robertson, L.A.; Armas, D.R.; Robbie, E.P.; Osmukhina, A.; Xu, D.; Li, H.; Solomon, S.D. A Phase 1 Dose-Escalation Study of the Cardiac Myosin Inhibitor Aficamten in Healthy Participants. JACC Basic Transl. Sci. 2022, 7, 763–775. [Google Scholar] [CrossRef]
- Ostrominski, J.W.; Guo, R.; Elliott, P.M.; Ho, C.Y. Cardiac Myosin Inhibitors for Managing Obstructive Hypertrophic Cardiomyopathy. JACC Heart Fail. 2023, 11, 735–748. [Google Scholar] [CrossRef]
- Canadian Agency for Drugs and Technologies in Health. CADTH Reimbursement Review Mavacamten (Camzyos); Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2023. [Google Scholar]
- Cytokinetics. Cytokinetics Announces New PDUFA Date for Aficamten in Obstructive Hypertrophic Cardiomyopathy; Cytokinetics: San Francisco, CA, USA, 2025. [Google Scholar]
- Aman, A.; Akram, A.; Akram, B.; Maham, M.; Bokhari, M.Z.; Akram, A.; Akram, S.; Yaqub, F. Efficacy of cardiac myosin inhibitors mavacamten and aficamten in hypertrophic cardiomyopathy: A systematic review and meta-analysis of randomised controlled trials. Open Heart 2025, 12, e003215. [Google Scholar] [CrossRef] [PubMed]
- Ramonfaur, D.; Gasperetti, A.; Blake, V.E.; Rivers, B.; Kassamali, A.A.; Kasper, E.K.; Barouch, L.A.; Wu, K.C.; Madrazo, J.A.; Carrick, R.T. Eighteen-Month Real-World Experience Using Mavacamten for Treatment of Obstructive Hypertrophic Cardiomyopathy in a Racially Diverse Population. J. Am. Heart Assoc. 2024, 13, e034069. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.M.Y.; Masri, A. Differentiating Cardiac Troponin Levels During Cardiac Myosin Inhibition or Cardiac Myosin Activation Treatments: Drug Effect or the Canary in the Coal Mine? Curr. Heart Fail. Rep. 2023, 20, 504–518. [Google Scholar] [CrossRef]
- Nair, A.; Xie, L.; Enciso, J.E.S. Myosin Inhibitors. J. Am. Coll. Cardiol. 2023, 81, 46–48. [Google Scholar] [CrossRef]
- Shah, S.J.; Rigolli, M.; Javidialsaadi, A.; Patel, R.B.; Khadra, S.; Goyal, P.; Little, S.; Wever-Pinzon, O.; Owens, A.T.; Skali, H.; et al. Cardiac Myosin Inhibition in Heart Failure with Normal and Supranormal Ejection Fraction. JAMA Cardiol. 2025, 10, 170. [Google Scholar] [CrossRef]
- Duqueney, E.; Reza, N.; Boakye, E.; Marzolf, A.; Hornsby, N.; De Feria, A.; Owens, A. Sex Differences in Characteristics of Patients with Symptomatic Obstructive Hypertrophic Cardiomyopathy Initiated on Mavacamten. J. Card. Fail. 2025, 31, 237. [Google Scholar] [CrossRef]
- Cresci, S.; Bach, R.G.; Saberi, S.; Owens, A.T.; Spertus, J.A.; Hegde, S.M.; Lakdawala, N.K.; Nilles, E.K.; Wojdyla, D.M.; Sehnert, A.J.; et al. Effect of Mavacamten in Women Compared with Men with Obstructive Hypertrophic Cardiomyopathy: Insights From EXPLORER-HCM. Circulation 2024, 149, 498–509. [Google Scholar] [CrossRef]
- Glavaški, M.; Velicki, L.; Vučinić, N. Hypertrophic Cardiomyopathy: Genetic Foundations, Outcomes, Interconnections, and Their Modifiers. Medicina 2023, 59, 1424. [Google Scholar] [CrossRef]
- Kinnear, C.; Said, A.; Meng, G.; Zhao, Y.; Wang, E.Y.; Rafatian, N.; Parmar, N.; Wei, W.; Billia, F.; Simmons, C.A.; et al. Myosin inhibitor reverses hypertrophic cardiomyopathy in genotypically diverse pediatric iPSC-cardiomyocytes to mirror variant correction. Cell Rep. Med. 2024, 5, 101520. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Mavacamten | Aficamten | |
|---|---|---|
| Class of Drug | Cardiac myosin inhibitor | Cardiac myosin inhibitor |
| Binding Site | Allosteric site distinct from aficamten | Same allosteric site as blebbistatin near pi-releasing backdoor of myosin |
| Type of Change in Myosin Confirmation | Stabilizes super relaxed state of myosin | Stabilizes weak actin binding pre-power stroke state |
| Key Mechanism of Action | Decreases the number of myosin heads available to form to crossbridge cycle with actin [22] | Decreases ATPase activity and sarcomere force through slowing phosphate (Pi) release [22] |
| Effect on Actin–Myosin Relationship | Reduces myosin–actin binding | Prevents the transition to strong actin-binding, force generating |
| Interaction with Blebbistatin | Does not compete with blebbistatin | Competes with blebbistatin (mutually exclusive binding) [22] |
| Half-Life and Steady State Confirmation | Long half-life (7–9 days), steady state in ~6 weeks [22] | Shorter half-life, reversible within 24–48 h, steady state in ~2 weeks [22] |
| Trial Name | Phase | Population | Method and Design | Primary Endpoints | Key Results/Outcomes |
|---|---|---|---|---|---|
| PIONEER-HCM [29] | 2 | Symptomatic obstructive HCM | Open-label, multicenter, mavacamten as monotherapy in cohort A and add to β-blockers in cohort B | Change in LVOT gradient at 12 weeks | Cohort A: LVOT gradient reduced from 103 mm Hg to 19 mm Hg (mean change: −89.5 mm Hg; p = 0.008); Peak VO2 increased by 3.5 mL/kg/min; LVEF decreased by 15%. Cohort B: LVOT gradient reduced from 86 mm Hg to 64 mm Hg (mean change: −25.0 mm Hg; p = 0.02); Peak VO2 increased by 1.7 mL/kg/min; LVEF decreased by 6% |
| MAVERICK-HCM [30] | 2 | Symptomatic non-obstructive HCM (NYHA II–III, LVEF ≥ 55%, NT-proBNP ≥ 300 pg/mL) | Randomized, double-blind, placebo controlled with initial 5 mg dose daily with 1 dose titration at week 6 | Safety and tolerability with secondary outcomes of NT-proBNP and hs-cTnI | Generally well tolerated and safe with 10% serious adverse effects of mavacamten group and 21% in placebo group. In addition, 5 participants stopped taking the drug due to LVEF reduction which was reversible within 4–12 weeks |
| EXPLORER-HCM [31] | 3 | Symptomatic obstructive HCM (NYHA II-III, LVOT gradient above 50 mmHg) | Double-blind RCT with placebo control and 30-week treatment period | Composite of ≥1.5 mL/kg/min increase in peak VO2 and ≥1 NYHA class improvement, or ≥3.0 mL/kg/min increase in peak VO2 without NYHA class worsening | Improved exercise capacity and symptoms, primary outcomes achieved in 37% mavacamten-treated versus 17% in placebo-treated patients (p = 0.0001). Peak VO2 increased by 1.4 mL/kg/min (p = 0.0006) in mavacamten group with significant improvements in NYHA class and quality of life measures. |
| Trial Name | Phase | Population | Method and Design | Primary Endpoints | Key Results/Outcomes |
|---|---|---|---|---|---|
| REDWOOD-HCM [33] | 2 | 41 patients with symptomatic nHCM patients with LVOT gradient ≤ 30 mmHg, LVEF ≥ 60%, NT-proBNP > 300 pg/mL | Open-label trial where patients received individualized aficamten doses of 5–15 mg once daily (titration based on echocardiography LVEF) for 10 weeks | Safety, tolerability and efficacy of aficamten (through measures of symptom burden and cardiac biomarkers) over 10 weeks | 55% showed NYHA class improvement, 29% became asymptomatic. NT-proBNP reduced by 56% (p = 0.001), hs-TnI by 22% (p = 0.005). Mean LVEF declined by 5.4%; 3 asymptomatic patients had transient LVEF < 50%. One fatal arrhythmia in a high-risk patient |
| SEQUOIA-HCM [32] | 3 | 282 patients with symptomatic obstructive HCM, LVEF ≥ 60%, LV wall thickness ≥ 15 mm | Double-blind, randomly assigned to take aficamten (5 mg, starting) or placebo for 24 weeks | Change in peak VO2 from baseline to 24 weeks | Peak Vo2 increased by 1.7 mL/kg/min vs. placebo (p = 0.0001). KCCQ-CSS improved by 12 vs. 5 points (p < 0.001). 49.3% achieved LVOT < 30 mmHg after Valsalva vs. 3.6% on placebo. Serious adverse events: 5.6% vs. 9.3% |
| Adverse Events | Mavacamten (%) | Aficamten (%) |
|---|---|---|
| Dizziness | 19% 2 | 4.2% 3 |
| Atrial Fibrillation | 19% 2 | 2.8% 3 |
| Death | 1.4% 1 | 0% 1 |
| LVEF < 50% | 10.6% 1 | 4.8% 1 |
| Heart Failure | 4.3% 1 | 0% 1 |
| LVEF Drop with Temporary [or Permanent] Discontinuation | 6.8% [1.9%] 1 | 0.5% [0%] 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savsin, H.; Tokarek, T. Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy. Biomedicines 2025, 13, 1619. https://doi.org/10.3390/biomedicines13071619
Savsin H, Tokarek T. Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy. Biomedicines. 2025; 13(7):1619. https://doi.org/10.3390/biomedicines13071619
Chicago/Turabian StyleSavsin, Helin, and Tomasz Tokarek. 2025. "Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy" Biomedicines 13, no. 7: 1619. https://doi.org/10.3390/biomedicines13071619
APA StyleSavsin, H., & Tokarek, T. (2025). Comprehensive Review: Mavacamten and Aficamten in Hypertrophic Cardiomyopathy. Biomedicines, 13(7), 1619. https://doi.org/10.3390/biomedicines13071619