


Spontaneous Necrosis of a High-Risk Bladder Tumor Under Immunotherapy for Concurrent Malignant Melanoma: Role of BRAF Mutations and PD-L1 Expression

 ,
,  , , , ,
, , , ,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Conceptual Design and Study Cohort
- The molecular evaluation of somatic BRAF gene mutations in the concurrently occurring bladder tumors, i.e., occurring under treatment with BRAF/MEK inhibitors for V600E positive, recurrent MM, as possible emerging drivers of carcinogenesis in BC;
- The assessment of PD-L1 immuno-expression in BC tissue as a predictor of therapeutic response to ICIs.
2.2. PD-L1-Targeted Immunohistochemistry Expression Analysis
2.2.1. Tissue Preparation and Staining Protocol
2.2.2. Interpretation and Scoring
2.2.3. Quality Control
2.3. BRAF Gene Mutation Analysis
2.3.1. Tissue Preparation and DNA Extraction
2.3.2. Mutation Detection Protocol
- p.V600A (c.1799T>C): Thymine replaced by cytosine at position 1799 of the coding DNA sequence (CDS), the substitution of valine (V) with alanine (A) at codon 600.
- p.V600D (c.1799_1800delTGinsAT): Deletion of thymine and guanine at positions 1799 and 1800 of the CDS, replaced by adenine and thymine, leading to a valine (V) to aspartic acid (D) substitution at codon 600.
- p.V600E (c.1799T>A): Thymine to adenine substitution at position 1799 of the CDS, resulting in valine (V) being replaced by glutamic acid (E) at codon 600.
- p.V600E (c.1799_1800delTGinsAA): Deletion of thymine and guanine at positions 1799 and 1800 of the CDS, replaced by two adenines, causing a valine (V) to glutamic acid (E) substitution at codon 600.
- p.V600G (c.1799T>G): Thymine to guanine substitution at position 1799 of the CDS, leading to valine (V) being replaced by glycine (G) at codon 600.
- p.V600K (c.1798_1799delGT>insAA): Deletion of guanine and thymine at positions 1798 and 1799 of the CDS, replaced by two adenines, resulting in valine (V) being substituted by lysine (K) at codon 600.
- p.V600M (c.1798G>A): Guanine to adenine substitution at position 1798 of the CDS, causing valine (V) to be replaced by methionine (M) at codon 600.
- p.V600R (c.1798_1799delGT>insAG): Deletion of guanine and thymine at positions 1798 and 1799 of the CDS, replaced by adenine and guanine, leading to valine (V) being substituted by arginine (R) at codon 600.
- p.K601E (c.1801A>G): Adenine to guanine substitution at position 1801 of the CDS, resulting in lysine (K) being replaced by glutamic acid (E) at codon 601.
- Assay qPCR BRAF Control V600E (3.125× concentration)
- Assay qPCR BRAF MUT V600E (3.125× concentration)
- Master Mix BRAF V600E (2.083× concentration)
- Standard WT BRAF V600E (104 copies/µL)
- Standard MUT 1% BRAF V600E (104 copies/µL)
- Deionized Water
- 7.
- Initial Denaturation: 95 °C for 3 min.
- 8.
- Denaturation: 95 °C for 10 s (50 cycles).
- 9.
- Annealing + Elongation (with fluorescence acquisition): 60 °C for 20 s (50 cycles).
2.3.3. Data Interpretation and Quality Controls
- FC vs. cutoff:
- Each run has an assigned cutoff FC value (often near 9–10, as adjusted by the WT standard).
- If a sample’s FC exceeds the cutoff FC, the sample is classified as WT.
- If a sample’s FC falls below the cutoff FC, the sample is considered mutated (provided the second criterion below is also met).
- ΔCt Confirmation:
- The difference (CtMSAMPLE − CtCWT-STANDARD) must be <13 cycles to confirm that the sample’s signal is truly within the quantifiable range of the assay.
- Samples with FC < cutoff, but (CtMSAMPLE − CtCWT-STANDARD) ≥ 13 are deemed below the LOD and thus effectively classified as WT (or unquantifiable mutation).
- Mutant allele fraction (%) = 100 × 1.93 − FC.
3. Results
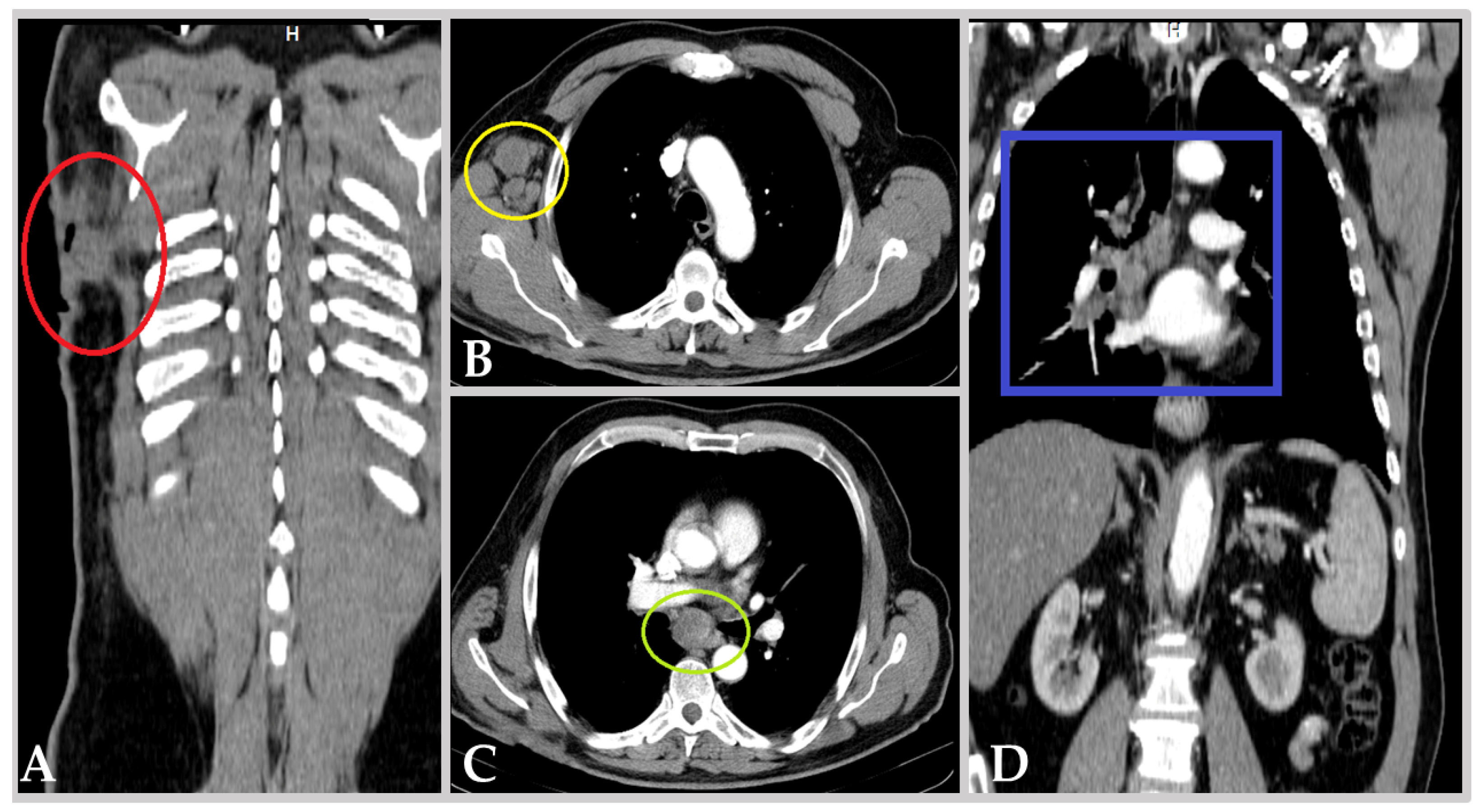
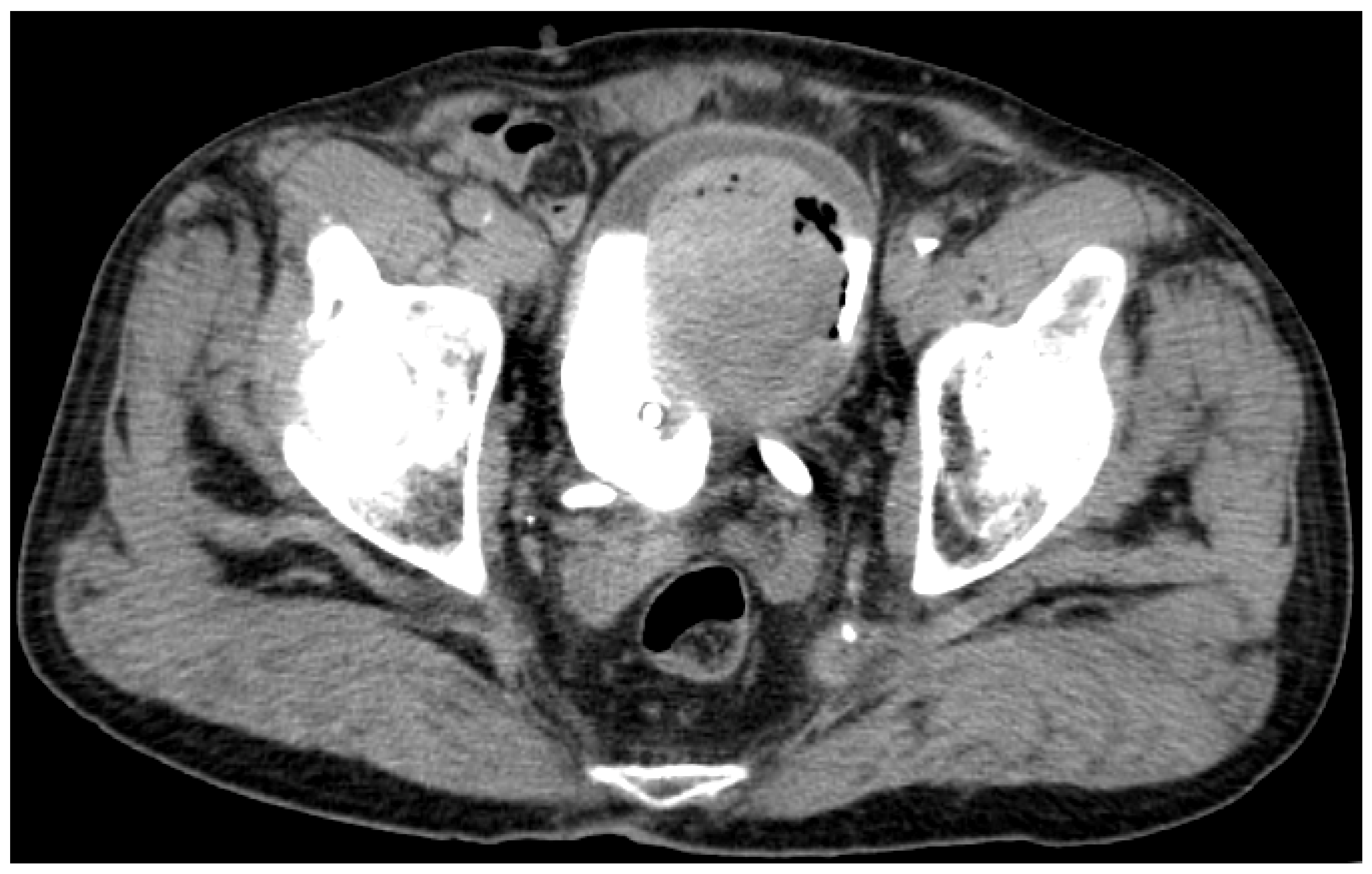
3.1. Clinical Background
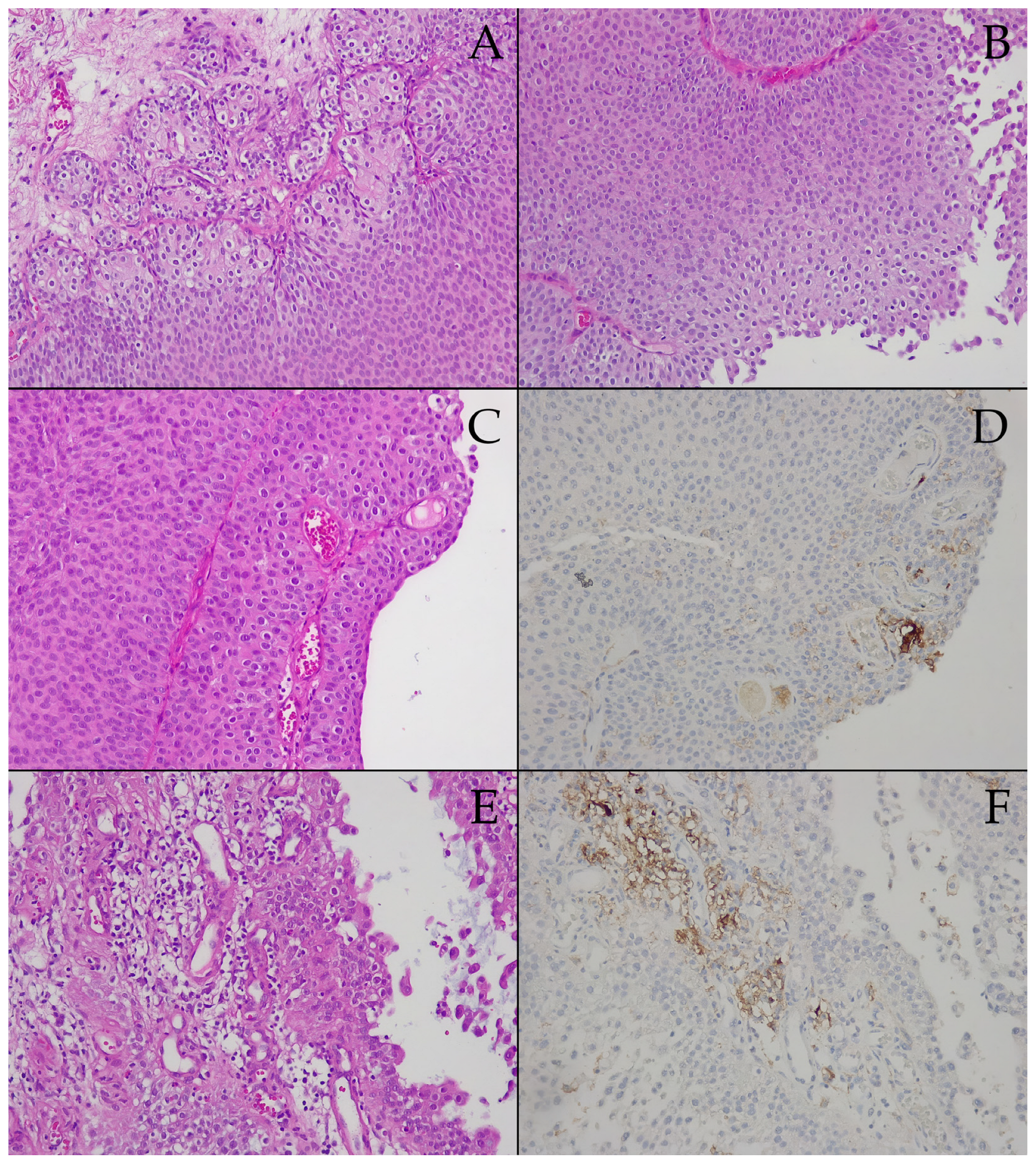
3.2. Prespontaneous Bladder Tumor Necrosis Tissue Samples
3.3. Postspontaneous Bladder Tumor Necrosis Tissue Samples
4. Discussion
4.1. Clinical Considerations
4.2. Molecular Oncogenesis and BRAF Gene Mutations Assessment
- Mutations mostly unrelated to clinical subtype—changes in chromosome 9 and/or the RAS family of proto-oncogenes (H-, K-, and N-RAS);
- Mutations related to specific grades/stages of the disease—fibroblast growth factor receptor 3 (FGFR3) mutations for low-grade noninvasive papillary urothelial BC; TP53 and RB1 alterations in muscle-invasive disease [40].
4.3. PD-L1 Expression and Treatment Response Prediction
- Tumor mutational burden (TMB)/neoantigen load: High TMB is associated with better response to ICIs, independent of PD-L1 status. Further genomic analysis would be required to determine whether these bladder tumors harbored a high TMB.
- Pre-existing immune activation: The prior anti-BRAF/MEK therapy may have primed the immune system, indirectly enhancing Nivolumab’s effectiveness. On the other hand, in correlation with the data previously presented, hinting at the possible pro-oncogenic role of BRAF/MEK inhibitors in BC, it may even be the case that, in fact, the interruption of anti-BRAF/MEK therapy was the real underlying cause of regression, not the initiation of immunotherapy.
- Microenvironment factors: The tumor’s immune microenvironment, including tumor-infiltrating lymphocytes and cytokine expression, may have contributed to an ICI-sensitive phenotype despite low PD-L1 expression.
- Vigilance for secondary malignancies in BRAF/MEK-treated patients: Clinicians should be aware of the potential for BC development during anti-BRAF/MEK therapy and ensure appropriate urological monitoring [63].
- Resistance to ICIs may evolve over time: The progressive decrease in PD-L1 expression in our case highlights the dynamic nature of tumor immune interactions, emphasizing the need for serial biomarker assessment in ICI-treated patients [55].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richters, A.; Aben, K.K.H.; Kiemeney, L.A.L.M. The Global Burden of Urinary Bladder Cancer: An Update. World J. Urol. 2020, 38, 1895–1904. [Google Scholar] [CrossRef]
- Bolla, S.R.; Odeluga, N.; Amraei, R.; Jetti, R. Histology, Bladder. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Mushtaq, J.; Thurairaja, R.; Nair, R. Bladder Cancer. Surgery 2019, 37, 529–537. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med. Sci. 2020, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.C.; Lee, S.; Lee, J.G.; Chen, H.; Zaleski, M.; Choi, W.; McConkey, D.J.; Wei, P.; Czerniak, B. Molecular Profile of Bladder Cancer Progression to Clinically Aggressive Subtypes. Nat. Rev. Urol. 2024, 21, 391–405. [Google Scholar] [CrossRef]
- Mariappan, P.; Fineron, P.; O’Donnell, M.; Gailer, R.M.; Watson, D.J.; Smith, G.; Grigor, K.M. Combining Two Grading Systems: The Clinical Validity and Inter-Observer Variability of the 1973 and 2004 WHO Bladder Cancer Classification Systems Assessed in a UK Cohort with 15 Years of Prospective Follow-Up. World J. Urol. 2021, 39, 425–431. [Google Scholar] [CrossRef]
- Grant, E.J.; Ozasa, K.; Preston, D.L.; Suyama, A.; Shimizu, Y.; Sakata, R.; Sugiyama, H.; Pham, T.-M.; Cologne, J.; Yamada, M.; et al. Effects of Radiation and Lifestyle Factors on Risks of Urothelial Carcinoma in the Life Span Study of Atomic Bomb Survivors. Radiat. Res. 2012, 178, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Moschini, M.; Zaffuto, E.; Karakiewicz, P.I.; Andrea, D.D.; Foerster, B.; Abufaraj, M.; Soria, F.; Mattei, A.; Montorsi, F.; Briganti, A.; et al. External Beam Radiotherapy Increases the Risk of Bladder Cancer When Compared with Radical Prostatectomy in Patients Affected by Prostate Cancer: A Population-Based Analysis. Eur. Urol. 2019, 75, 319–328. [Google Scholar] [CrossRef]
- Minami, T.; Fujita, K.; Hashimoto, M.; Nishimoto, M.; Adomi, S.; Banno, E.; Nozawa, M.; Nose, K.; Yoshimura, K.; Inada, M.; et al. External Beam Radiotherapy Combination Is a Risk Factor for Bladder Cancer in Patients with Prostate Cancer Treated with Brachytherapy. World J. Urol. 2023, 41, 1317–1321. [Google Scholar] [CrossRef]
- Chou, W.H.; McGregor, B.; Schmidt, A.; Carvalho, F.L.F.; Hirsch, M.S.; Chang, S.L.; Kibel, A.; Mossanen, M. Cyclophosphamide-Associated Bladder Cancers and Considerations for Survivorship Care: A Systematic Review. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Spiess, P.E.; Czerniak, B. Dual-Track Pathway of Bladder Carcinogenesis: Practical Implications. Arch. Pathol. Lab. Med. 2006, 130, 844–852. [Google Scholar] [CrossRef] [PubMed]
- EAU Guidelines on Non-Muscle-Invasive Bladder Cancer—Uroweb. Available online: https://uroweb.org/guidelines/non-muscle-invasive-bladder-cancer/chapter/pathological-staging-and-classification-systems (accessed on 1 February 2025).
- Kim, L.H.C.; Patel, M.I. Transurethral Resection of Bladder Tumour (TURBT). Transl. Androl. Urol. 2020, 9, 3056–3072. [Google Scholar] [CrossRef]
- Meeks, J.J.; Al-Ahmadie, H.; Faltas, B.M.; Taylor, J.A.; Flaig, T.W.; DeGraff, D.J.; Christensen, E.; Woolbright, B.L.; McConkey, D.J.; Dyrskjøt, L. Genomic Heterogeneity in Bladder Cancer: Challenges and Possible Solutions to Improve Outcomes. Nat. Rev. Urol. 2020, 17, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Capaldo, B.J.; Roller, D.; Axelrod, M.J.; Koeppel, A.F.; Petricoin, E.F.; Slingluff, C.L.; Weber, M.J.; Mackey, A.J.; Gioeli, D.; Bekiranov, S. Data from: Systems Analysis of Adaptive Responses to MAP Kinase Pathway Blockade in BRAF Mutant Melanoma. PLoS ONE 2016, 10, e0138210. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Kobayashi, M.; Onozawa, M.; Watanabe, S.; Nagashima, T.; Tamura, K.; Kubo, Y.; Ikeda, A.; Ochiai, K.; Michishita, M.; Bonkobara, M.; et al. Establishment of a BRAF V595E-Mutant Canine Prostate Cancer Cell Line and the Antitumor Effects of MEK Inhibitors against Canine Prostate Cancer. Vet. Comp. Oncol. 2023, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Gedon, J.; Kehl, A.; Aupperle-Lellbach, H.; von Bomhard, W.; Schmidt, J.M. BRAF Mutation Status and Its Prognostic Significance in 79 Canine Urothelial Carcinomas: A Retrospective Study (2006–2019). Vet. Comp. Oncol. 2022, 20, 449–457. [Google Scholar] [CrossRef]
- Aeschlimann, L.; Kehl, A.; Guscetti, F.; Posthaus, C.; Aupperle-Lellbach, H.; Rottenberg, S.; de Brot, S. Effective Detection of BRAF Mutation in Canine Urothelial and Prostate Carcinomas Using Immunohistochemistry. Vet. Comp. Oncol. 2024, 22, 295–302. [Google Scholar] [CrossRef]
- Decker, B.; Parker, H.G.; Dhawan, D.; Kwon, E.M.; Karlins, E.; Davis, B.W.; Ramos-Vara, J.A.; Bonney, P.L.; McNiel, E.A.; Knapp, D.W.; et al. Homologous Mutation to Human BRAF V600E Is Common in Naturally Occurring Canine Bladder Cancer—Evidence for a Relevant Model System and Urine-Based Diagnostic Test. Mol. Cancer Res. 2015, 13, 993–1002. [Google Scholar] [CrossRef]
- Younis, A.; Gribben, J. Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions. Immuno 2024, 4, 186–210. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination Therapy with Immune Checkpoint Inhibitors (ICIs); a New Frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M. Biomarkers of Response to Checkpoint Inhibitors beyond PD-L1 in Lung Cancer. Mod. Pathol. 2022, 35, 66–74. [Google Scholar] [CrossRef]
- Mehrsai, A.; Mansoori, D.; Taheri Mahmoudi, M.; Sina, A.; Seraji, A.; Pourmand, G.H. A Comparison between Clinical and Pathologic Staging in Patients with Bladder Cancer. Urol. J. 2004, 1, 85–89. [Google Scholar]
- Compérat, E.; Oszwald, A.; Wasinger, G.; Hansel, D.E.; Montironi, R.; van der Kwast, T.; Witjes, J.A.; Amin, M.B. Updated Pathology Reporting Standards for Bladder Cancer: Biopsies, Transurethral Resections and Radical Cystectomies. World J. Urol. 2022, 40, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Boegemann, M.; Krabbe, L.-M. Prognostic Implications of Immunohistochemical Biomarkers in Non-Muscle-Invasive Blad Cancer and Muscle-Invasive Bladder Cancer. Mini Rev. Med. Chem. 2020, 20, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and Risk Factors of Urothelial Bladder Cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Oszczudlowski, M.; Dobruch, J. Prediction of Progression to Muscle-Invasive Disease in Patients with High-Risk Bladder Cancer. Transl. Androl. Urol. 2018, 7, 749–751. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, S.; Alfred Witjes, J. Long-Term Cancer-Specific Survival in Patients with High-Risk, Non-Muscle-Invasive Bladder Cancer and Tumour Progression: A Systematic Review. Eur. Urol. 2011, 60, 493–500. [Google Scholar] [CrossRef]
- Richterstetter, M.; Wullich, B.; Amann, K.; Haeberle, L.; Engehausen, D.G.; Goebell, P.J.; Krause, F.S. The Value of Extended Transurethral Resection of Bladder Tumour (TURBT) in the Treatment of Bladder Cancer. BJU Int. 2012, 110 Pt 2, E76–E79. [Google Scholar] [CrossRef] [PubMed]
- Lavery, H.J.; Stensland, K.D.; Niegisch, G.; Albers, P.; Droller, M.J. Pathological T0 Following Radical Cystectomy with or without Neoadjuvant Chemotherapy: A Useful Surrogate. J. Urol. 2014, 191, 898–906. [Google Scholar] [CrossRef]
- Engilbertsson, H.; Aaltonen, K.E.; Björnsson, S.; Kristmundsson, T.; Patschan, O.; Rydén, L.; Gudjonsson, S. Transurethral Bladder Tumor Resection Can Cause Seeding of Cancer Cells into the Bloodstream. J. Urol. 2015, 193, 53–57. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.; Lee, D.; Figueiredo, A.; Hermanns, T.; Wild, P.; Komosa, M.; Lau, I.; Mistry, M.; Nunes, N.M.; Price, A.J.; et al. Combined Genetic and Epigenetic Alterations of the TERT Promoter Affect Clinical and Biological Behavior of Bladder Cancer. Int. J. Cancer 2019, 144, 1676–1684. [Google Scholar] [CrossRef]
- Tran, L.; Xiao, J.-F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in Bladder Cancer Biology and Therapy. Nat. Rev. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular Biology of Bladder Cancer: New Insights into Pathogenesis and Clinical Diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhu, Y.; Ji, A.; Zhang, Q.; Liao, G. Mining TCGA Database for Tumor Mutation Burden and Their Clinical Significance in Bladder Cancer. Biosci. Rep. 2020, 40, BSR20194337. [Google Scholar] [CrossRef] [PubMed]
- Boulalas, I.; Zaravinos, A.; Delakas, D.; Spandidos, D.A. Mutational Analysis of the BRAF Gene in Transitional Cell Carcinoma of the Bladder. Int. J. Biol. Markers 2009, 24, 17–21. [Google Scholar] [CrossRef]
- Isaacson, A.L.; Guseva, N.V.; Bossler, A.D.; Ma, D. Urothelial Carcinoma with an NRF1-BRAF Rearrangement and Response to Targeted Therapy. Mol. Case Stud. 2019, 5, a003848. [Google Scholar] [CrossRef]
- Merrill, N.M.; Vandecan, N.M.; Day, K.C.; Palmbos, P.L.; Day, M.L.; Udager, A.M.; Merajver, S.D.; Soellner, M.B. MEK Is a Promising Target in the Basal Subtype of Bladder Cancer. Oncotarget 2020, 11, 3921–3932. [Google Scholar] [CrossRef]
- Wu, J.H.; Cohen, D.N.; Rady, P.L.; Tyring, S.K. BRAF Inhibitor-Associated Cutaneous Squamous Cell Carcinoma: New Mechanistic Insight, Emerging Evidence for Viral Involvement and Perspectives on Clinical Management. Br. J. Dermatol. 2017, 177, 914–923. [Google Scholar] [CrossRef]
- Cho, N.-Y.; Choi, M.; Kim, B.-H.; Cho, Y.-M.; Moon, K.C.; Kang, G.H. BRAF and KRAS Mutations in Prostatic Adenocarcinoma. Int. J. Cancer 2006, 119, 1858–1862. [Google Scholar] [CrossRef]
- Burger, M.; Denzinger, S.; Hammerschmied, C.; Tannapfel, A.; Maderstorfer, A.; Wieland, W.F.; Hartmann, A.; Stoehr, R. Mitogen-Activated Protein Kinase Signaling Is Activated in Prostate Tumors but Not Mediated by B-RAF Mutations. Eur. Urol. 2006, 50, 1102–1109. [Google Scholar] [CrossRef]
- Sommerer, F.; Hengge, U.R.; Markwarth, A.; Vomschloss, S.; Stolzenburg, J.-U.; Wittekind, C.; Tannapfel, A. Mutations of BRAF and RAS Are Rare Events in Germ Cell Tumours. Int. J. Cancer 2005, 113, 329–335. [Google Scholar] [CrossRef]
- Nagy, A.; Balint, I.; Kovacs, G. Frequent Allelic Changes at Chromosome 7q34 but Lack of Mutation of the BRAF in Papillary Renal Cell Tumors. Int. J. Cancer 2003, 106, 980–981. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Summersgill, B.; Spendlove, H.E.; Huddart, R.; Houlston, R.; Shipley, J. Activating Mutations and/or Expression Levels of Tyrosine Kinase Receptors GRB7, RAS, and BRAF in Testicular Germ Cell Tumors. Neoplasia 2005, 7, 1047–1052. [Google Scholar] [CrossRef]
- Ueda, M.; Toji, E.; Nunobiki, O.; Izuma, S.; Okamoto, Y.; Torii, K.; Noda, S. Mutational Analysis of the BRAF Gene in Human Tumor Cells. Hum. Cell 2008, 21, 13–17. [Google Scholar] [CrossRef]
- Liu, H.; Ye, T.; Yang, X.; Lv, P.; Wu, X.; Zhou, H.; Lu, H.; Tang, K.; Ye, Z. Predictive and Prognostic Role of PD-L1 in Urothelial Carcinoma Patients with Anti-PD-1/PD-L1 Therapy: A Systematic Review and Meta-Analysis. Dis. Markers 2020, 2020, 8375348. [Google Scholar] [CrossRef]
- Bi, X.; Zhao, X.; Wang, J.; Li, Y.; Li, J.; Jiang, Q.; Yang, W.; Liu, X.; Liu, H.; Liu, L. Radiomics-Based Nomogram to Predict Tumor Mutation Burden in Muscle-Invasive Bladder Cancer. Diagnostics 2022, 12, 929. [Google Scholar] [CrossRef]
- Wang, L.; Wu, C.; Huang, S.; Liu, H.; Zhang, H.; Tang, W. Radiomics Signatures for Predicting Response to Immunotherapy in Bladder Cancer. Front. Oncol. 2023, 13, 1173922. [Google Scholar]
- Powles, T.; Assaf, Z.J.; Degaonkar, V.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; Albers, P.; Castellano, D.; Nishiyama, H.; et al. Updated Overall Survival by Circulating Tumor DNA Status from the Phase 3 IMvigor010 Trial: Adjuvant Atezolizumab Versus Observation in Muscle-Invasive Urothelial Carcinoma. Eur. Urol. 2024, 85, 114–122. [Google Scholar] [CrossRef]
- Herranz, R.; Oto, J.; Plana, E.; Fernández-Pardo, Á.; Cana, F.; Martínez-Sarmiento, M.; Vera-Donoso, C.D.; España, F.; Medina, P. Circulating Cell-Free DNA in Liquid Biopsies as Potential Biomarker for Bladder Cancer: A Systematic Review. Cancers 2021, 13, 1448. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Skog, J. Exosome-Based Liquid Biopsies in Cancer: Opportunities and Challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B. The Role of Exosomes in Bladder Cancer. Pharmaceutics 2022, 14, 2027. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Dynamic Regulation of PD-L1 Expression and Its Impact on Response to Immune Checkpoint Blockade. J. Clin. Oncol. 2021, 39, 467–479. [Google Scholar]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Ptashkin, R.N.; Kuo, F.; Riaz, N.; Harris, L.; Hollmann, T.; et al. Tumor Mutation Burden as an Independent Predictor of Response to Immunotherapy Across Cancer Types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumor-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential Macrophage Programming in the Tumor Microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Sibaud, V.; Lamant, L.; Maisongrosse, V.; Delord, J.P. BRAF Inhibitors and Secondary Malignancies: A Systematic Review. Cancer Treat. Rev. 2020, 86, 102033. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.; Castellano, D.; Duran, I.; Mironov, S.; et al. Clinical and Biomarker-Based Predictors of Response to PD-L1 Inhibition in Bladder Cancer. Lancet Oncol. 2021, 22, 897–910. [Google Scholar]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Parikh, A.; Lee, J.L.; Rha, S.Y.; Gupta, S.; et al. IMvigor130: PD-L1 Expression as a Predictor of Response to Atezolizumab in Advanced Urothelial Carcinoma. Eur. Urol. 2022, 81, 123–134. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Procedure | Findings | Concomitant Treatment 1 |
|---|---|---|---|
| 12 July 2019 | TURBT | - pTaG2 high grade; - muscularis propria absent. | Dabrafenib + Trametinib |
| 2 June 2020 | TURBT | - pTaG2 high grade, but with residual tumor; - muscularis propria present. | Nivolumab |
| 9 July 2020 | TURBT | - pT1G2 high grade (biopsy), i.e., large residual tumor. | Nivolumab |
| 30 September 2020 | Cistoscopy | - complete spontaneous tumor necrosis. | Nivolumab |
| 1 February 2021 | TURBT | - pT1G2 high grade, with focal squamous features; - muscularis propria absent. | Nivolumab |
| 23 March 2021 | Radical Cystectomy | - pTaG2 high grade, “early urothelial carcinoma”, R0; - N0 (34 lymph nodes); - incidental prostatic adenocarcinoma, GS 3 + 3 = 6, pT2a | Nivolumab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condoiu, C.; Musta, M.; Cumpanas, A.A.; Bardan, R.; Dema, V.; Zara, F.; Suciu, C.S.; Dumitru, C.-S.; Ciucurita, A.; Dumache, R.; et al. Spontaneous Necrosis of a High-Risk Bladder Tumor Under Immunotherapy for Concurrent Malignant Melanoma: Role of BRAF Mutations and PD-L1 Expression. Biomedicines 2025, 13, 377. https://doi.org/10.3390/biomedicines13020377
Condoiu C, Musta M, Cumpanas AA, Bardan R, Dema V, Zara F, Suciu CS, Dumitru C-S, Ciucurita A, Dumache R, et al. Spontaneous Necrosis of a High-Risk Bladder Tumor Under Immunotherapy for Concurrent Malignant Melanoma: Role of BRAF Mutations and PD-L1 Expression. Biomedicines. 2025; 13(2):377. https://doi.org/10.3390/biomedicines13020377
Chicago/Turabian StyleCondoiu, Cristian, Mihael Musta, Alin Adrian Cumpanas, Razvan Bardan, Vlad Dema, Flavia Zara, Cristian Silviu Suciu, Cristina-Stefania Dumitru, Andreea Ciucurita, Raluca Dumache, and et al. 2025. "Spontaneous Necrosis of a High-Risk Bladder Tumor Under Immunotherapy for Concurrent Malignant Melanoma: Role of BRAF Mutations and PD-L1 Expression" Biomedicines 13, no. 2: 377. https://doi.org/10.3390/biomedicines13020377
APA StyleCondoiu, C., Musta, M., Cumpanas, A. A., Bardan, R., Dema, V., Zara, F., Suciu, C. S., Dumitru, C.-S., Ciucurita, A., Dumache, R., Ismail, H., & Novacescu, D. (2025). Spontaneous Necrosis of a High-Risk Bladder Tumor Under Immunotherapy for Concurrent Malignant Melanoma: Role of BRAF Mutations and PD-L1 Expression. Biomedicines, 13(2), 377. https://doi.org/10.3390/biomedicines13020377