Abstract
Obesity, characterized by excessive body fat, is closely linked to endoplasmic reticulum (ER) stress, leading to insulin resistance and type 2 diabetes. Inflammatory pathways like c-Jun N-terminal kinase (JNK) worsen insulin resistance, impacting insulin signaling. Moreover, ER stress plays a substantial role in cancer, influencing tumor cell survival and growth by releasing factors like vascular endothelial growth factor (VEGF). The unfolded protein response (UPR) is pivotal in this process, offering both pro-survival and apoptotic pathways. This review offers an extensive exploration of the sophisticated connection between ER stress provoked by obesity and its role in both the onset and advancement of cancer. It delves into the intricate interplay between oncogenic signaling and the pathways associated with ER stress in individuals who are obese. Furthermore, this review sheds light on potential therapeutic strategies aimed at managing ER stress induced by obesity, with a focus on addressing cancer initiation and progression. The potential to alleviate ER stress through therapeutic interventions, which may encompass the use of small molecules, FDA-approved medications, and gene therapy, holds great promise. A more in-depth examination of pathways such as UPR, ER-associated protein degradation (ERAD), autophagy, and epigenetic regulation has the potential to uncover innovative therapeutic approaches and the identification of predictive biomarkers.
1. Introduction
Cancer has emerged as the second leading cause of death globally, stimulating extensive research in biomedical science to unravel its origins, tissue formation, and metastasis. It is responsible for approximately 14.1 million new cases and 8.2 million deaths annually [1]. Despite substantial investments and research efforts, cancer continues to pose challenges. Nevertheless, advancements at the cellular and molecular levels have enhanced our understanding of its etiology and spread, leading to continuous progress in cancer treatments [2,3]. While established risk factors for cancer include genetic predisposition, ionizing radiation, tobacco use, infections, unhealthy diet, alcohol consumption, sedentary lifestyle, and environmental exposures, obesity has emerged as a significant risk factor for various types of cancer. The prevalence of cancer is expected to rise due to the increasing occurrence of risk factors, particularly obesity and metabolic syndrome [3].
Adipose tissue normally plays a crucial role in regulating metabolic balance. However, metabolic disorders like obesity and type 2 diabetes often manifest with persistent inflammation and dysfunction in adipose tissue. Obesity, characterized by abnormal or excessive fat accumulation in adipose tissues, is considered a chronic inflammatory condition [4,5]. Previous studies have indicated that obesity-associated adipose tissue is exposed to various stressful conditions, including ER stress [6,7]. Interestingly, aberrant activation of ER stress sensors and their downstream signaling pathways have emerged as important regulators of tumor growth, metastasis, and response to cancer therapies such as chemotherapy, targeted therapies, and immunotherapy [8,9]. This review provides an in-depth examination of how ER stress triggered by obesity contributes to the initiation and advancement of cancer, along with the interplay between oncogenic signaling and various ER stress-related pathways in obese individuals. Furthermore, we explore potential therapeutic strategies aimed at addressing ER stress as a means of managing obesity. Additionally, we share our perspectives on approaches and future directions for modulating ER stress in the clinical treatment of obesity.
2. Endoplasmic Reticulum Structure and Functions
In 1902, Emilio Veratti made the initial discovery of the endoplasmic reticulum (ER) in muscle fibers, initially referred to as the sarcoplasmic reticulum, which was later recognized as comparable to the ER in other cells. Subsequently, Keith Porter visualized the ER using electron microscopy and named it the endoplasmic reticulum [10].
ER is a dynamic and expansive cellular structure with multiple functions, including calcium storage, protein synthesis, and lipid metabolism.
The structure of ER is unique, comprising an outer nuclear envelope and a continuous membranous system known as the rough ER, along with a network of tubules and sheets known as the smooth ER (Figure 1). The dynamic morphology of the ER plays a significant role in protein biosynthesis. Additionally, the smooth ER is involved in lipid metabolism and enzyme detoxification [11].
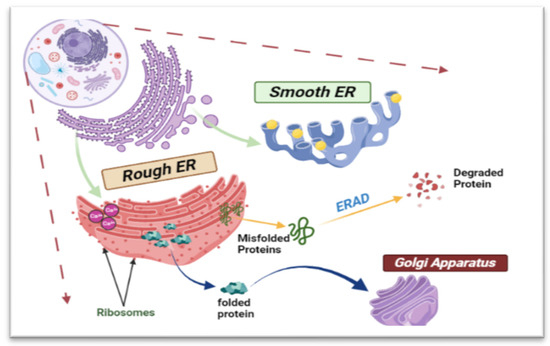
Figure 1.
Schematic diagram showing the structure and functions of the ER. This complicated cellular organelle consists of interconnected tubules and sacs, with the rough endoplasmic reticulum (RER) housing ribosomes on its surface and the smooth endoplasmic reticulum (SER) lacking ribosomes. Misfolded proteins will be degraded through ER-associated degradation (ERAD).
2.1. Calcium Storage and Metabolism
The ER, a complex membranous network, crucially manages calcium storage and release, with calcium ions (Ca2+) as vital regulators of cellular processes [12]. The sarcoplasmic reticulum within the ER serves as the primary compartment for storing releasable calcium. The specialized region on the ER membrane, the sarcoplasmic/endoplasmic reticulum calcium ATPase pump (SERCA), plays a key role in maintaining cellular homeostasis by transporting Ca2+ from the cytosol to the sarcoplasmic reticulum [13,14]. Upon receiving a signal, functional membrane proteins, such as inositol trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs), are triggered to release Ca2+. The general design of these storage facilities appears to be planned to minimize any subsequent alterations, hence preserving the functional chaperones of luminal proteins [12].
Following the release of Ca2+, the ions should be removed to restore the resting calcium concentration to prepare for the next signal (Figure 2). This can be accomplished through active transport using the SERCA pump, passive transport via the plasma membrane calcium pumps, and sequestration into organelles like the mitochondria and the Golgi apparatus.
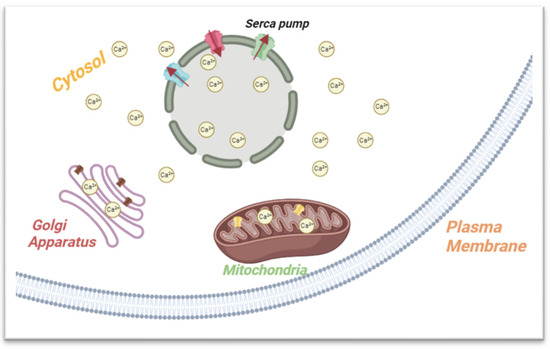
Figure 2.
Summary of the ER’s role in calcium storage and metabolism. The ER membrane features SERCA, a crucial regulator maintaining cellular balance by moving calcium ions (Ca2+) from the cytosol to the ER, preserving low cytoplasmic calcium levels. After calcium release, restoration of resting levels occurs through SERCA, passive plasma membrane pumps, and organelle sequestration (e.g., mitochondria, Golgi).
ER calcium storage significantly impacts cancer development, and understanding the complexities of Ca2+ transport and signaling in cancer cells holds promise for novel therapeutic strategies targeting calcium homeostasis [13]. Key points on ER calcium storage and cancer: (1) ER Ca2+ transporters: their aberrations in cancer contribute significantly to regulating ER Ca2+ stores; (2) Ca2+ signaling: this involves receptor isoform expression and downstream effector landscape3, often exploited in malignancies; (3) Ca2+-based therapeutic interventions: insights into ER Ca2+ transporters offer potential for therapeutic interventions; (4) mitochondria-associated membranes (MAMs): connectivity through MAMs is a primary mechanism for Ca2+ entry into non-excitable cells, modulated by intracellular Ca2+; (5) oncogenes and tumor suppressors: Ca2+ action centrality in carcinogenesis is linked to several oncogenes and tumor suppressors [13,14].
2.2. Protein Synthesis and Folding
The ER’s membranous structures are involved in various cellular processes, with their primary functions being centered around the proper production, modification, and quality control of proteins. The ER’s several structural domains—each of which is linked to a particular function—enable it to produce cytosolic, integral membrane, and secreted proteins (Figure 3) [11].
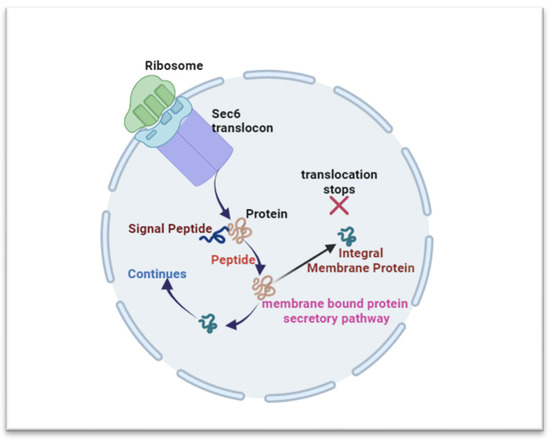
Figure 3.
Overview of the ER’s involvement in protein synthesis and folding. The ER’s structural domains support protein production, with ribosomes on its surface. The canonical pathway anchors mRNA for translation initiation in the cytosol. Proteins are translated in the cytosol and continue in the ER via Sec6 translocon. A peptidase aids signal peptide cleavage, releasing the protein (cytosolic, integral membrane, and secreted proteins) into the ER lumen.
The ER-driven protein synthesis involves ribosome arrangement, guided by the canonical pathway and co-transitional mRNA anchoring [11,15]. Translation initiates in the cytosol and shifts to the ER membrane through the Sec6 translocon, enabling the polypeptide’s ER entry [11,16]. Following translation, a peptidase cleaves the signal peptide, releasing the nascent protein into the ER lumen [11,17]. Proteins take distinct paths; integral membrane proteins halt translocation, while those for secretion enter the ER for modifications. Post-translation, proteins undergo activity, stability, and specificity-altering modifications [18,19]. Proteins for secretion undergo proper folding and modifications, guided by chaperones and enzymes, while those for ER function initiate folding. Misfolding triggers ER-associated degradation (ERAD) or retention in the ER [11,19].
ER protein synthesis plays a crucial role in cancer development, and its dysregulation can lead to the activation of the unfolded protein response UPR and the promotion of cancer cell apoptosis, as elaborated in the subsequent section. Understanding the mechanisms of ER protein synthesis and its connection with cancer development can help in the development novel therapeutic strategies [15,16].
2.3. Lipid Metabolism
ER lipid metabolism is a vital cellular process essential for maintaining cell structure and function [20]. It serves as the primary site for lipid synthesis, modification, and regulation, encompassing diverse molecules like fats, phospholipids, and sterols [20]. These lipids play key roles in cell membranes, energy storage, signaling, and cellular process regulation. The ER is centrally involved in lipid synthesis and distribution throughout the cell [21].
Dysregulated lipid metabolism is a key metabolic shift in cancer, marked by heightened lipid synthesis, storage, and uptake, contributing to cancer progression [17]. Tumor cells exhibit increased de novo lipogenesis, fatty acid uptake, and oxidation for energy and lipid accumulation. This dysregulation impacts membrane composition, gene expression, signaling pathways, and cellular functions, influencing cancer initiation and progression [18]. Lipid droplets (LDs) are crucial in cancer, serving as storage sites and regulating fatty acid flow and energy balance. Reprogramming lipid metabolism, including neo-lipogenesis and fatty acid oxidation changes, is critical in tumorigenesis [19] (C). Targeting the aberrant lipid metabolism presents a potential avenue for more effective cancer treatments.
2.4. Regulation of Lipid Metabolism
Lipid metabolism regulation encompasses pathways, such as the inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and pancreatic ER kinase-like ER kinase (PERK) pathways, which are pivotal elements of the UPR. These pathways play indispensable roles in maintaining cellular lipid equilibrium and shielding against biotoxicity [20]. IRE1, serving as a key UPR sensor, orchestrates cellular responses to the ER stress by controlling genes associated with lipid biosynthesis and metabolism [21]. PERK, another UPR sensor, influences lipid metabolism by modulating global protein synthesis and facilitating the activation of the transcription factor 4 (ATF4)-mediated transcription of genes linked to lipid regulation [22]. ATF6, the third UPR sensor, relocates to the nucleus during ER stress, activating genes responsible for lipid synthesis and adaptive responses [23]. Collectively, these pathways ensure the precise lipid balance within cells, guarding against cellular dysfunction stemming from lipid imbalances [20].
2.4.1. IRE1
The IRE1α/X-box binding protein 1 (XBP1) pathway activates lipid-related genes like Lipin genes, Osbp, Pecr, Lss, and Gpat4, influencing lipid synthesis [24]. XBP1 primarily impacts lipogenesis gene translation, crucial during liver transitions between fasting and refeeding [20]. Liver-specific Xbp1 knockout disrupts fatty acid and sterol synthesis, lowering plasma triglyceride levels. XBP1 contributes to ER expansion, influencing phosphatidylcholine and phosphatidylethanolamine synthesis. It aids in metabolic adaptation during fasting and ketogenic diets, with tissue-specific effects. Regulated IRE1α-dependent decay (RIDD) adds complexity to lipid metabolism, with outcomes shaped by the interplay between XBP1 and RIDD activities, leading to context-dependent effects [25,26,27,28,29,30].
2.4.2. PERK
PERK signaling, crucial for lipid balance, involves downstream components (eIF2α, ATF4, CHOP) and sensitive Insig-1, initiating lipogenesis. Absence of PERK in mammary cells reduces milk fat. Depleting Insig-1 in WT cells prompts SREBP processing via PERK and eIF2α, hindered in PERK KO cells. Comparable outcomes occur during hypotonic and ER stress [31,32]. In HCMV-infected cells, Insig-1 and SREBP1 activation by PERK operates independently of eIF2α phosphorylation. Overexpressing BiP in mice diminishes ER stress and SREBP-1c cleavage, reinforcing the UPR’s role [33,34,35]. Liver-specific eIF2α dephosphorylation impacts gluconeogenesis and lipid synthesis, affecting fasting-induced low blood sugar and high-fat-diet-induced liver fat. Transient activation of PERK-induced lipo- and glycogenic pathways is observed. Phosphorylated eIF2α boosts ATF4 translation, influencing TG homeostasis. ATF4 KO mice on a high-carbohydrate diet show reduced hepatic and serum TG levels, improved glucose and insulin tolerance, and reduced lipogenic enzymes. ATF4 deficiency protects against diet-induced liver steatosis by reducing hepatic lipogenesis without impacting TG secretion and FA oxidation, shielding against fructose-induced hepatic hypertriglyceridemia [36,37].
2.4.3. ATF6
ATF6 regulates lipid metabolism independently of XBP1, enhancing choline kinase activity and phosphatidylcholine synthesis in NIH-3T3 cells. This effect, similar to XBP1, involves post-transcriptional or post-translational mechanisms [38]. ATF6 overexpression also increases Acacb and Fasn expression, enhancing fatty acid biosynthesis. In response to glucose deprivation, ATF6 inhibits cholesterol synthesis by interacting with processed SREBP2, suppressing lipogenesis in HepG2 cells. Direct activation by sphingolipid intermediates triggers ATF6’s transcriptional program related to lipids. ATF6’s role in lipid metabolism is linked to PPARα, promoting fatty acid oxidation [39]. Moreover, ATF6 deficiency or dnATF6 overexpression in mice results in liver steatosis, while ATF6 activation prevents triglyceride accumulation and enhances fatty acid oxidation. ATF6 interacts with PPARα/RXRα heterodimers, crucial for their transcriptional activity in the liver, especially during fasting. Furthermore, ATF6 plays a more critical role in promoting fatty acid oxidation than synthesis in the liver [40,41].
2.5. ER Homeostasis and ER Stress
ER is crucial for cellular functions, focusing on protein synthesis and folding. Disruptions, like unfolded proteins, trigger intricate responses: translation inhibition, selective degradation, and increased chaperones. Restoring ER function is vital for cell viability; any imbalance can lead to demise. ER stress, impacting functions like lipid and calcium metabolism, leads to abnormalities (Table 1). Persistent accumulation of misfolded or unfolded proteins overwhelms cellular quality control, resulting in ER stress. Exploring ER stress deepens understanding of strategies used by cells to preserve protein-folding balance, revealing their remarkable adaptability in challenging environments [42].

Table 1.
ER Function in normal cells and the effects of ER Stress.
ER stress arises from unfolded or misfolded proteins in the ER lumen, triggered by infections, lipid accumulation, glucose deprivation, disturbed redox balance, or calcium regulation (Table 2) [5]. Persistent buildup of irregular proteins leads to cell apoptosis [6]. The body initiates the UPR to regulate new protein production and restore ER capacity, with ATF6, PERK, and IRE1, trans-membrane proteins in the lipid metabolism, influencing the UPR [6,45,46]. These proteins convey messages from the ER to the cytoplasm or nucleus, activating three pathways: (i) suppressing protein translation to prevent additional unfolded proteins; (ii) inducing ER molecular chaperone genes for protein folding; and (iii) activating ERAD to reduce unfolded protein accumulation. Unsuccessful attempts result in cellular imbalance and apoptosis [6,46,47,48].
Table 2.
Common factors inducing endoplasmic reticulum (ER) stress.
Obese adipose tissue exhibits enhanced adipocyte hypertrophy and hyperplasia, along with chronic inflammation involving inflammatory cell infiltration and cytokine network activation [6,49,50,51]. Recent studies link obesity to ER stress, which can trigger or result from obesity [52,53,54,55]. ER stress induced by obesity can lead to insulin resistance and type 2 diabetes, particularly in adipose tissues [56,57]. Elevated cytokines and free fatty acids in obesity signal c-JNK activation, implicated in inflammatory responses. In type 2 diabetes, JNK activation is associated with insulin resistance [58]. ER stress in obesity downregulates insulin receptor substrate-1 (IRS1) tyrosine phosphorylation (enhancing insulin signal inhibition) and upregulates serine IRS1 phosphorylation (inhibiting insulin signal) [57] (Figure 4). The IRE1 pathway, including XBP1, signals UPR-related gene transcription for protein folding and ER chaperones, limiting the ER stress response in cases of elevated XBP1 levels [57].
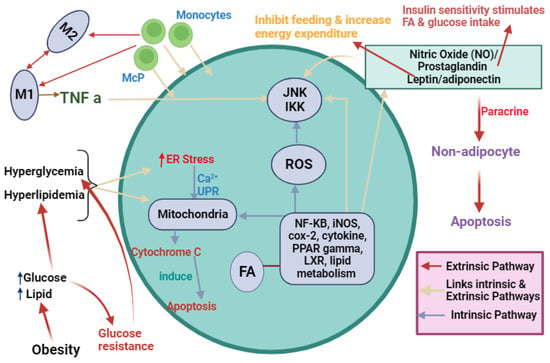
Figure 4.
Outline of the key factors affecting adipocyte inflammation and metabolism leading to ER stress. Obesity induces hyperglycemia and hyperlipidemia, triggering ER stress through an intrinsic pathway leading to apoptosis. Another pathway (extrinsic) involving nitric oxide (NO), prostaglandin, and leptin/adiponectin activates JKK and IKK signaling, inhibiting feeding and increasing energy expenditure. Simultaneously, it triggers nuclear factor-κB (NF-KB), inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (cox-2), cytokines, peroxisome proliferator-activated receptors (PPAR) gamma, liver X receptor (LXR), and lipid metabolism, culminating in paracrine signaling and adipocyte apoptosis. Monocyte differentiation into macrophages releases TNF-α, activating JNK and IKK pathways. This intricate network orchestrates adipocyte responses, linking extracellular cues to inflammation, metabolic changes, and apoptosis.
2.6. ER Stress and Cancer
Interestingly, emerging research has highlighted the refined connection between ER stress and cancer development. The complex interplay between ER stress and cancer involves both protective mechanisms aimed at restoring cellular homeostasis and pathways that can promote tumor growth and survival (Table 3). In tumor cells, ER stress may lead to the restoration of homeostasis and the creation of an environment that is favorable for the survival and growth of the tumor by releasing growth factors, cytokines, and angiogenic factors [59,60]. ER stress has the potential not only to impede cell apoptosis through its cytotoxic actions but also to promote tumor growth.
Table 3.
Cancer types and associated ER stress markers expression.
Unfolded protein response (UPR) induction is brought on by ER stress. Tumor cells exhibit both apoptotic and adaptive UPR. By increasing the secretion of vascular endothelial growth factor (VEGF), the adaptive action of UPR triggers anti-apoptotic NF-B, which blocks p53-dependent apoptotic signals and stimulates angiogenic activity. Moreover, IRE1, ATF6, and PERK, which are in the ER, are three ER-stress-signaling branches that are involved in carcinogenesis. The down-signaling XBP1 and IRE1 have a role in the development of cancer [76]. The progression of cancer is also aided by the PERK/eIF2/ATF4 branch of the ER stress pathway [77]. Separately, the ER-resident chaperone calreticulin, which is connected to both immunogenic cell death and the localization of calreticulin on the surfaces of tumor cells, has been localized to the cell surface in tumor cells. This connection might be linked to the production of ER stress in tumor cells.
Pancreatic cancer, a highly aggressive malignancy, has garnered significant attention due to its relationship with ER stress. The endoplasmic reticulum is a vital cellular organelle responsible for protein synthesis and folding, and disruptions in its normal functions can trigger ER stress. In pancreatic cancer, a combination of factors such as nutrient deprivation, oxidative stress, and an intense demand for protein synthesis can overwhelm the ER, leading to ER stress. This stress response has been implicated in various aspects of pancreatic cancer progression, including tumor initiation, growth, metastasis, and therapy resistance. The consequences of ER stress in pancreatic cancer involve complex cellular mechanisms, such as the UPR, which can either promote cancer cell survival or contribute to its demise. Pancreatic cancer proposes a very active UPR, which makes it resistant to therapy and promotes disease progression [50].
The three UPR receptors, IRE1, ATF6, and PERK, remain evident in pancreatic cancer. Glucose-regulated protein 78 (GRP78), also recognized as binding immunoglobulin protein (BiP), is an ER chaperone found in the ER’s lumen. It hinders the activation of IRE1, PERK, and ATF6 by binding to their ER luminal segments. Accumulation of misfolded proteins within the ER, observed during instances of ER stress, leads to the separation of GRP78. This, in turn, triggers the activation of IRE1, PERK, and ATF6, along with their subsequent signaling pathways.
2.6.1. IRE1
Inositol-requiring enzyme 1 (IRE1), a vital mediator of the UPR in the endoplasmic reticulum (ER), plays a crucial role in cancer development [51], regulating processes like phosphoinositide signaling lipids and macrophage growth. Key points on IRE1’s link to cancer include IRE1α and angiogenesis; IRE1α is a key regulator of angiogenesis, promoting blood vessel growth in tumors [52]. Moreover, IRE1 regulates phosphoinositide-signaling lipids crucial for cancer cell growth and survival [52].
2.6.2. PERK
PERK, a pancreatic ER kinase, is vital in the unfolded protein response (UPR) within the endoplasmic reticulum (ER) and is linked to cancer development [53]. Despite this association, the direct link between PERK and cancer remains incompletely understood. Key points about PERK and its connection to cancer include PERK’s implication in regulating ER stress, which has links to cancer development [53]. ER stress can trigger the UPR, exhibiting both adaptive and cytotoxic effects on tumor cells1. The precise link between PERK and cancer is not fully elucidated; however, ER stress and the UPR can have both adaptive and cytotoxic effects on tumor cells. ER stress may either enhance tumor growth or induce apoptosis [54]. Moreover, obesity is associated with the development of ER stress, contributing to various diseases, including cancer [55]. However, the specific relationship between PERK and obesity remains unclear.
2.6.3. ATF6
ATF6 plays a crucial role in cancer development and has been found to regulate carcinoma progression through mTOR or PERK signaling pathways [47]. In the context of cancer and obesity, ATF6 has been implicated in the development of obesity-related cancers. Some key points about ATF6 and its association with cancer and obesity include the fact that ATF6 has been found to promote cancer cell survival and proliferation by inducing endoplasmic reticulum stress, which, in turn, promotes autophagy and confers cancer cells’ resistance to chemotherapy treatment [47]. ER stress can induce the UPR, which can have both adaptive and cytotoxic effects on tumor cells [47]. Furthermore, obesity-induced ER stress causes chronic inflammation in adipose tissue, and the expression levels of ER stress markers were significantly up-regulated in obese mice. This suggests that ATF6, which plays a role in ER stress, may be involved in the development of obesity-related cancers. Additionally, ATF6 has been found to play a significant role in the tumor microenvironment, promoting the growth of cancer cells and the development of metastases [56].
2.6.4. GRP78
GRP78 is associated with obesity-related diseases and is also implicated in various aspects of cancer development, making it a potential target for therapeutic interventions in both obesity and cancer [57]. In the context of cancer, GRP78 is involved in several aspects of cancer development, including tumor survival, chemoresistance, angiogenesis, and metastasis. Many tumor cells overexpress GRP78 on the outer plasma membrane, and its expression has been correlated with tumor resistance, greater risk for cancer recurrence, and a decrease in patient survival [64]. Therefore, GRP78 at the cell surface has been proposed as a promising target for cancer therapeutics [64]. Additionally, covalent inhibition of GRP78 has been shown to disconnect the transduction of ER stress signals to inflammation and lipid accumulation in diet-induced obese mice, suggesting its potential role in anti-obesity effects [58].
3. Targeting Obesity Induced ER Stress Mediator for Cancer Therapy
Therapeutics targeting ER stress represent an emerging approach in the treatment of various diseases, particularly those associated with protein misfolding, inflammation, and cellular stress. Since ER is responsible for protein folding and lipid synthesis [78,79], when it becomes overwhelmed due to factors like an excessive buildup of unfolded or misfolded proteins, it triggers a stress response known as the UPR. Prolonged or severe ER stress can lead to cell dysfunction and cell death, contributing to the pathogenesis of several diseases.
Strategies directed at mitigating ER stress could offer a novel path for therapeutic interventions in addressing the disorder. For instance, there have been encouraging outcomes observed in treating the condition using methods like small molecule compounds, FDA-approved substances, and gene therapy, all with the goal of re-establishing ER balance. Furthermore, delving deeper into clinical trials and examining each distinct pathway involved, such as the UPR, ERAD, and autophagy, in the context of maintaining or restoring ER equilibrium, holds the potential to uncover innovative therapeutic approaches and possibly identify biomarkers that could predict clinical outcomes [79].
In the complex landscape of obesity-induced cancer, a multitude of promising therapeutic targets emerge, each holding the potential to alleviate the heightened cancer risk associated with obesity. Chief among these targets is IRE1α, a central component of the UPR pathway. Recent studies have yielded compelling evidence of IRE1α’s pivotal role in orchestrating the survival and proliferation of cancer cells within the obese microenvironment. Notably, a groundbreaking study conducted in this context concluded that targeting IRE1α not only disrupts the adaptive responses of cancer cells to ER stress but also enhances the efficacy of oncolytic therapies. This finding, marked by increased cancer cell vulnerability and improved treatment outcomes, underscores the critical importance of IRE1α as a therapeutic node [80].
Furthermore, another study was conducted that aims to decipher the role of UPR signaling in melanoma, a notoriously apoptosis-resistant skin cancer. It investigates whether heightened UPR pro-survival signaling results from increased ER stress due to genetic and environmental factors or whether it is actively induced by common melanoma-associated mutations. The study suggests utilizing ER stress to tilt UPR signaling toward apoptosis but acknowledges the need for precision to avoid undesirable outcomes. It advocates a multifaceted approach, combining agents inhibiting cytoprotective UPR signaling with those inducing ER stress, potentially adding metabolic interventions and immune activators to improve melanoma therapy. Ultimately, the study envisions harnessing ER stress for more effective melanoma treatment, with ongoing trials expected to further inform these strategies [81].
Expanding our scope beyond IRE1α, the roles of PERK and ATF6, also integral to the UPR, have garnered attention as promising targets in the battle against obesity-induced cancer. These pathways play pivotal roles in regulating the cellular stress response, and their precise modulation holds the potential to disrupt cancer cell resilience.
Moreover, molecular chaperones like GRP78/BiP, autophagy regulators, and ROS modulators are under scrutiny for their ability to hinder cancer cell adaptation and survival. Meanwhile, the intricate metabolic changes occurring during ER stress in the context of obesity are subjects of intense investigation, with researchers seeking ways to exploit these alterations for therapeutic benefit.
In parallel, the epigenetic landscape within obesity-related cancer initiation is being explored, opening new horizons for the modulation of epigenetic regulators as potential targets for intervention. The collective exploration of these diverse targets not only promises to revolutionize cancer treatment strategies but also offers hope to individuals navigating the complex interplay between obesity and cancer. As researchers continue to unravel the intricacies of these pathways and their potential for therapeutic manipulation, the future holds the promise of more effective and personalized approaches to countering obesity-induced cancer.
Author Contributions
Conceptualization, L.A.-M.; writing—original draft preparation, J.A. and H.A.-J.; writing—review and editing, J.A., H.A.-J., S.A. and L.A.-M.; supervision, L.A.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding, and The APC was funded by The Qatar National Library (QNL).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization. Cancer. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 3 February 2022).
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217. [Google Scholar] [CrossRef]
- Staff AMCaN. Risk of Dying from Cancer Continues to Drop at an Accelerated Pace: American Cancer Society. 2022. Available online: https://www.cancer.org/research/acs-research-news/facts-and-figures-2022.html (accessed on 25 January 2024).
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Zhang, A.M.; Wellberg, E.A.; Kopp, J.L.; Johnson, J.D. Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab. J. 2021, 45, 285–311. [Google Scholar] [CrossRef]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef]
- Krzysztof, M.; Katarzyna, K.; Jolanta, S.-K.; Christine, W. Excessive Endoplasmic Reticulum Stress Correlates with Impaired Mitochondrial Dynamics, Mitophagy and Apoptosis. Liver and Adipose Tissue, but Not in Muscles in EMS Horses. Int. J. Mol. Sci. 2018, 19, 165. [Google Scholar]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- You, M.; Xie, Z.; Zhang, N.; Zhang, Y.; Xiao, D.; Liu, S.; Zhuang, W.; Li, L.; Tao, Y. Signaling pathways in cancer metabolism: Mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 196. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, Y.; Wang, Y.; Wang, L.; Zhang, L.; Zhao, Z.; Li, S. Lactobacillus plantarum S9 alleviates lipid profile, insulin resistance, and inflammation in high-fat diet-induced metabolic syndrome rats. Sci. Rep. 2022, 12, 15490. [Google Scholar] [CrossRef]
- Zhai, X.; Sterea, A.M.; El Hiani, Y. Lessons from the endoplasmic reticulum Ca2+ Transporters—A Cancer Connection. Cells 2020, 9, 1536. [Google Scholar] [CrossRef]
- Pedriali, G.; Rimessi, A.; Sbano, L.; Giorgi, C.; Wieckowski, M.R.; Previati, M.; Pinton, P. Regulation of endoplasmic reticulum–mitochondria Ca2+ transfer and its importance for anti-cancer therapies. Front. Oncol. 2017, 7, 180. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational regulations in response to endoplasmic reticulum stress in cancers. Cells 2020, 9, 540. [Google Scholar] [CrossRef]
- Fu, X.; Cui, J.; Meng, X.; Jiang, P.; Zheng, Q.; Zhao, W.; Chen, X. Endoplasmic reticulum stress, cell death and tumor: Association between endoplasmic reticulum stress and the apoptosis pathway in tumors. Oncol. Rep. 2021, 45, 801–808. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Wu, J.; Ren, Z. Lipid droplets: A cellular organelle vital in cancer cells. Cell Death Discov. 2023, 9, 254. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Allen, D.; Seo, J. ER Stress activates the TOR pathway through Atf6. J. Mol. Signal. 2018, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.-C.; Cui, H.; Mason, B.L.; Mahgoub, M.; Bookout, A.L.; Hana, G.Y.; Perello, M.; Elmquist, J.K.; Repa, J.J.; Zigman, J.M.; et al. Chronic social defeat stress disrupts regulation of lipid synthesis. J. Lipid Res. 2010, 51, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Maduka, I.; Neboh, E.; Ufelle, S. The relationship between serum cortisol, adrenaline, blood glucose and lipid profile of undergraduate students under examination stress. Afr. Health Sci. 2015, 15, 131–136. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Tirasophon, W.; Kaufman, R.J. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999, 7, 293–300. [Google Scholar]
- Basaiawmoit, R.V.; Rattan, S.I. Cellular stress and protein misfolding during aging. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols; Humana Press: Totowa, NJ, USA, 2010; Volume 648, pp. 107–117. [Google Scholar]
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The stress of protein misfolding: From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704. [Google Scholar] [CrossRef]
- Kiani, A.K.; Dhuli, K.; Donato, K.; Aquilanti, B.; Velluti, V.; Matera, G.; Iaconelli, A.; Connelly, S.T.; Bellinato, F.; Gisondi, P.; et al. Main nutritional deficiencies. J. Prev. Med. Hyg. 2022, 63 (Suppl. S3), E93. [Google Scholar]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Haeri, M.; Knox, B.E. Endoplasmic reticulum stress and unfolded protein response pathways: Potential for treating age-related retinal degeneration. J. Ophthalmic Vis. Res. 2012, 7, 45–59. [Google Scholar] [PubMed]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol. Cell 2019, 74, 877–890.e6. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic reticulum stress and oxidative stress: A vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Choi, J.-A.; Song, C.-H. Insights into the role of endoplasmic reticulum stress in infectious diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharmacother. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Palade, G.E. The endoplasmic reticulum. J. Biophys. Biochem. Cytol. 1956, 2, 85. [Google Scholar] [CrossRef]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.R.G.; Singleton, D.C.; Buffa, F.; Abramczyk, O.; Phadwal, K.; Li, J.-L.; Simon, A.K.; Murray, J.T.; Harris, A.L. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013, 449, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging roles of activating transcription factor (ATF) family members in tumourigenesis and immunity: Implications in cancer immunotherapy. Genes Dis. 2022, 9, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An emerging role for the unfolded protein response in pancreatic cancer. Cancers 2021, 13, 261. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological targeting of IRE1 in cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Tripathi, Y.B.; Pandey, V. Obesity and endoplasmic reticulum (ER) stresses. Front. Immunol. 2012, 3, 240. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J. ER stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2021, 43, 97–109. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Pan, D.; Yang, Y.; Nong, A.; Tang, Z.; Li, Q.X. GRP78 activity moderation as a therapeutic treatment against obesity. Int. J. Environ. Res. Public Health 2022, 19, 15965. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Fan, N.; Zhang, X.; Ngo, F.Y.; Zhao, J.; Zhao, W.; Huang, M.; Li, D.; Wang, Y.; Rong, J. Covalent inhibition of endoplasmic reticulum chaperone GRP78 disconnects the transduction of ER stress signals to inflammation and lipid accumulation in diet-induced obese mice. Elife 2022, 11, e72182. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Targeting endoplasmic reticulum signaling pathways in cancer. Acta Oncol. 2012, 51, 822–830. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PLoS ONE 2009, 4, e8008. [Google Scholar] [CrossRef]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Delie, F.; Petignat, P.; Cohen, M. GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J. Oncol. 2012, 2012, 468615. [Google Scholar] [CrossRef]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Overley-Adamson, B.; Artlett, C.M.; Stephens, C.; Sassi-Gaha, S.; Weis, R.D.; Thacker, J.D. Targeting the unfolded protein response, XBP1, and the NLRP3 inflammasome in fibrosis and cancer. Cancer Biol. Ther. 2014, 15, 452–462. [Google Scholar] [CrossRef][Green Version]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Déry, M.-A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Zhang, Y.; Tseng, C.-C.; Tsai, Y.-L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS ONE 2013, 8, e80071. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Zhu, G.; Pfaffenbach, K.; Kanel, G.; Stiles, B.; Lee, A.S. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene 2014, 33, 4997–5005. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Li, Z.; Li, H.; Song, H.; Bao, C.; Wei, J.; Cheng, L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer 2010, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, A.O.; Simpson, K.E.; Oyelere, S.F.; Nur, M.; Ngule, C.M.; Owoyemi, B.C.D.; Ayarick, V.A.; Oyelami, F.F.; Obaleye, O.; Esoe, D.-P.; et al. Unveiling the dark side of glucose-regulated protein 78 (GRP78) in cancers and other human pathology: A systematic review. Mol. Med. 2023, 29, 122. [Google Scholar] [CrossRef]
- Lee, H.K.; Xiang, C.; Cazacu, S.; Finniss, S.; Kazimirsky, G.; Lemke, N.; Lehman, N.L.; Rempel, S.A.; Mikkelsen, T.; Brodie, C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-Oncol. 2008, 10, 236–243. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wen, W.; Luo, J. Targeting endoplasmic reticulum stress as an effective treatment for alcoholic pancreatitis. Biomedicines 2022, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, G.; Martinotti, S.; Ranzato, E. Endoplasmic reticulum stress and cancer: Could unfolded protein response be a druggable target for cancer therapy? Int. J. Mol. Sci. 2023, 24, 1566. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.S.; Lovat, P.E.; Haass, N.K. Induction of endoplasmic reticulum stress as a strategy for melanoma therapy: Is there a future? Melanoma Manag. 2014, 1, 127–137. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).