
Xylosyltransferase-Deficiency in Human Dermal Fibroblasts Induces Compensatory Myofibroblast Differentiation and Long-Term ECM Reduction

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. CRISPR/Cas9-Based KO in Human Dermal Fibroblasts
2.3. Fluorescence-Based Cell Sorting and Cell Separation
2.4. TA Cloning
2.5. siRNA-Based Double-Knockdown (dKD)
2.6. RNA Extraction, Reverse Transcription and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.7. Xyloslytransferase Activity Assay
2.8. Galactosyltransferase Activity Assay
2.9. Determination of the Sulfated GAG (sGAG) Concentration in Fibroblasts
2.10. Western Blot
2.11. Bicinchoninic Acid Assay
2.12. Determination of the Pro-MMP1 Concentration in the Cell Culture Supernatants
2.13. Determination of the TGFβ1 Concentration in Cell Culture Supernatants
2.14. Determination of Cell Viability and Caspase Activity in Fibroblasts
2.15. Quantitative Senescence Assay
2.16. Determination of the Interleukin 6 (IL6) Concentration in Cell Culture Supernatants
2.17. Determination of the Intracellular Ca2+ Concentration of Fibroblasts
2.18. Inhibition of the TGFβ Receptor Kinase and Focal Adhesion-Associated Kinase (FAK) in Fibroblasts
2.19. Immunofluorescence
2.20. Statistical Analysis
3. Results
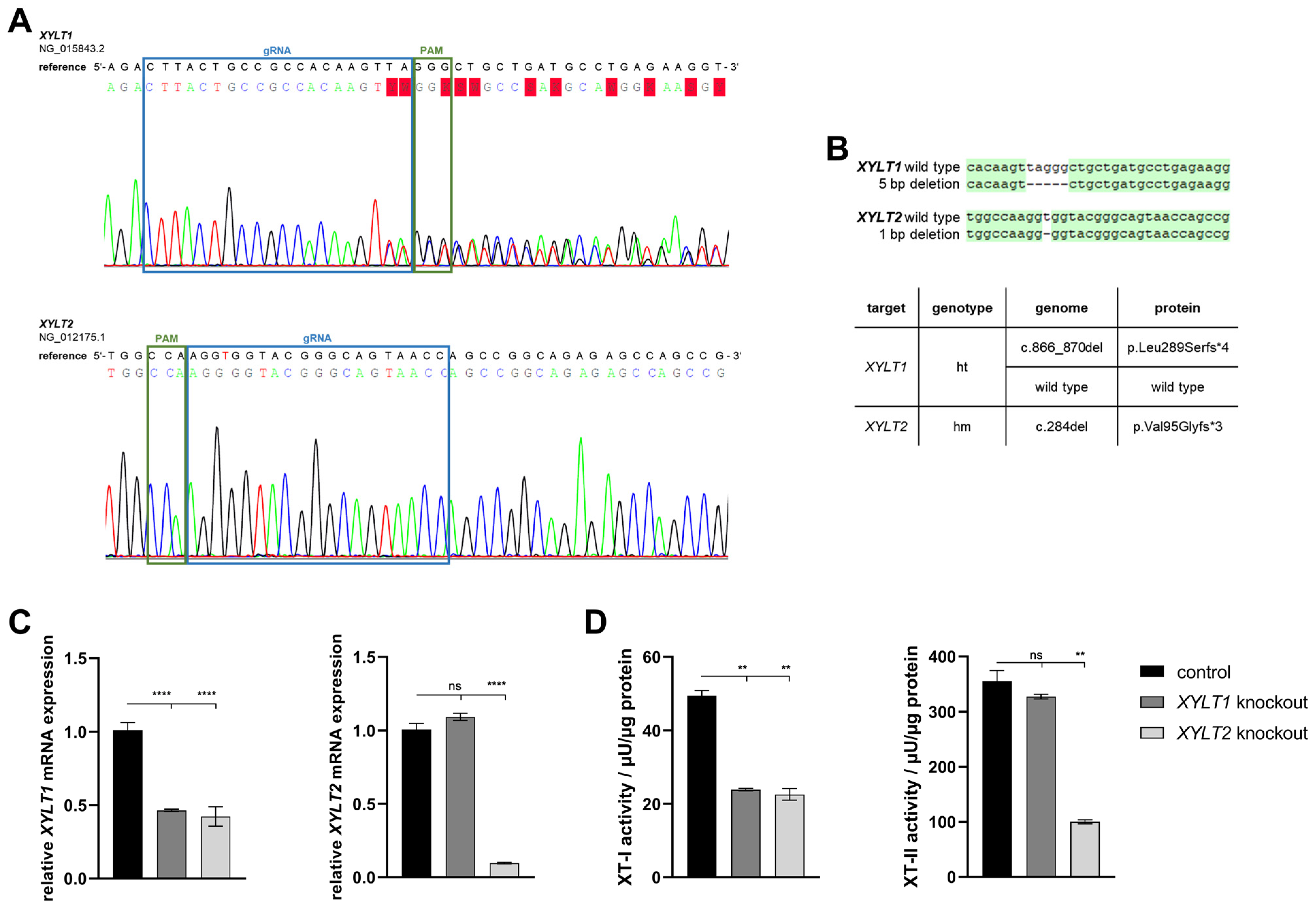
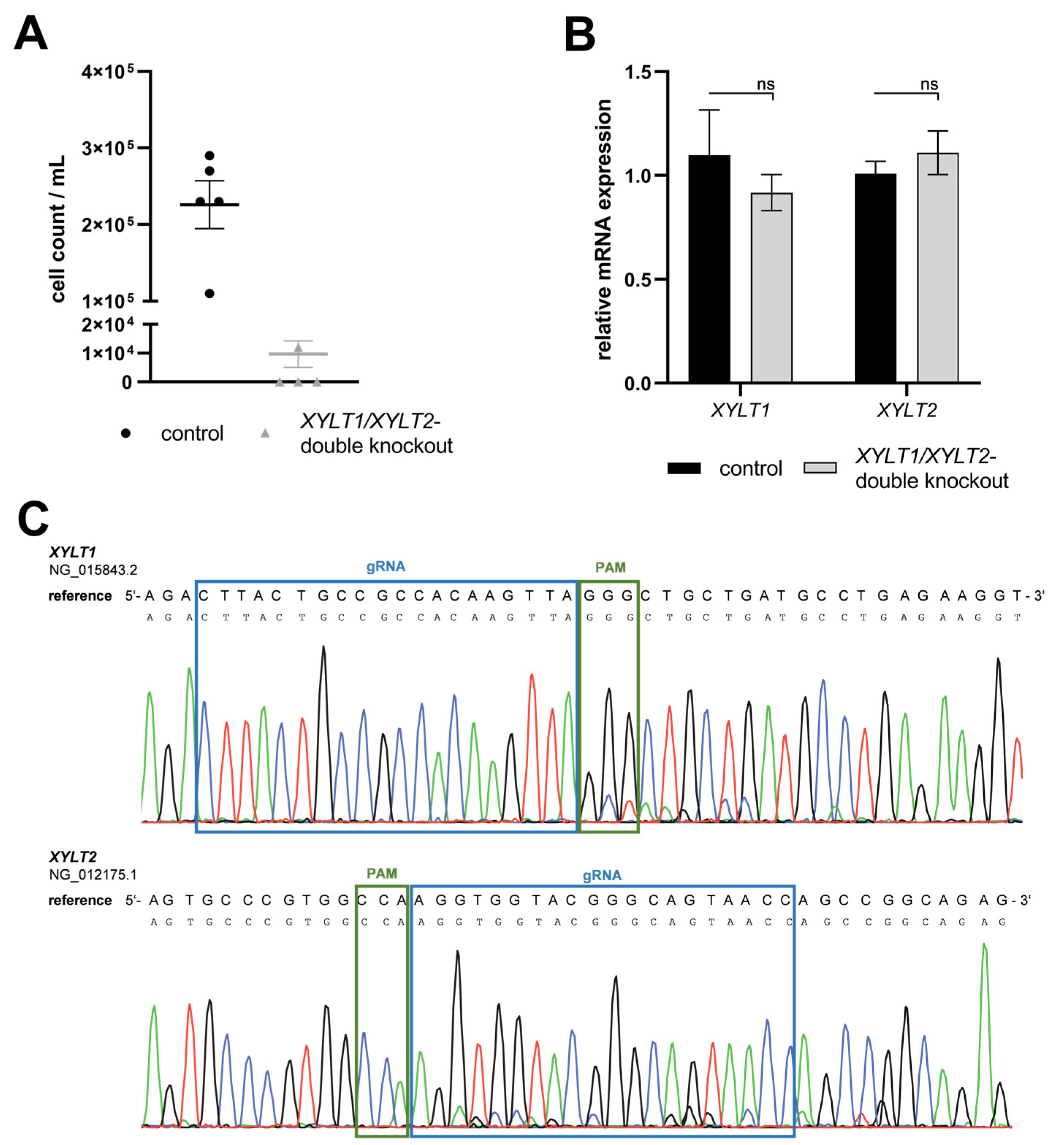
3.1. Generation of Separated CRISPR/Cas9-Mediated XYLT1 and XYLT2 KOs in Human Dermal Fibroblasts and Their Initial Characterization
3.2. Establishment of a Simultaneous XYLT1 and XYLT2 KO in Dermal Fibroblasts
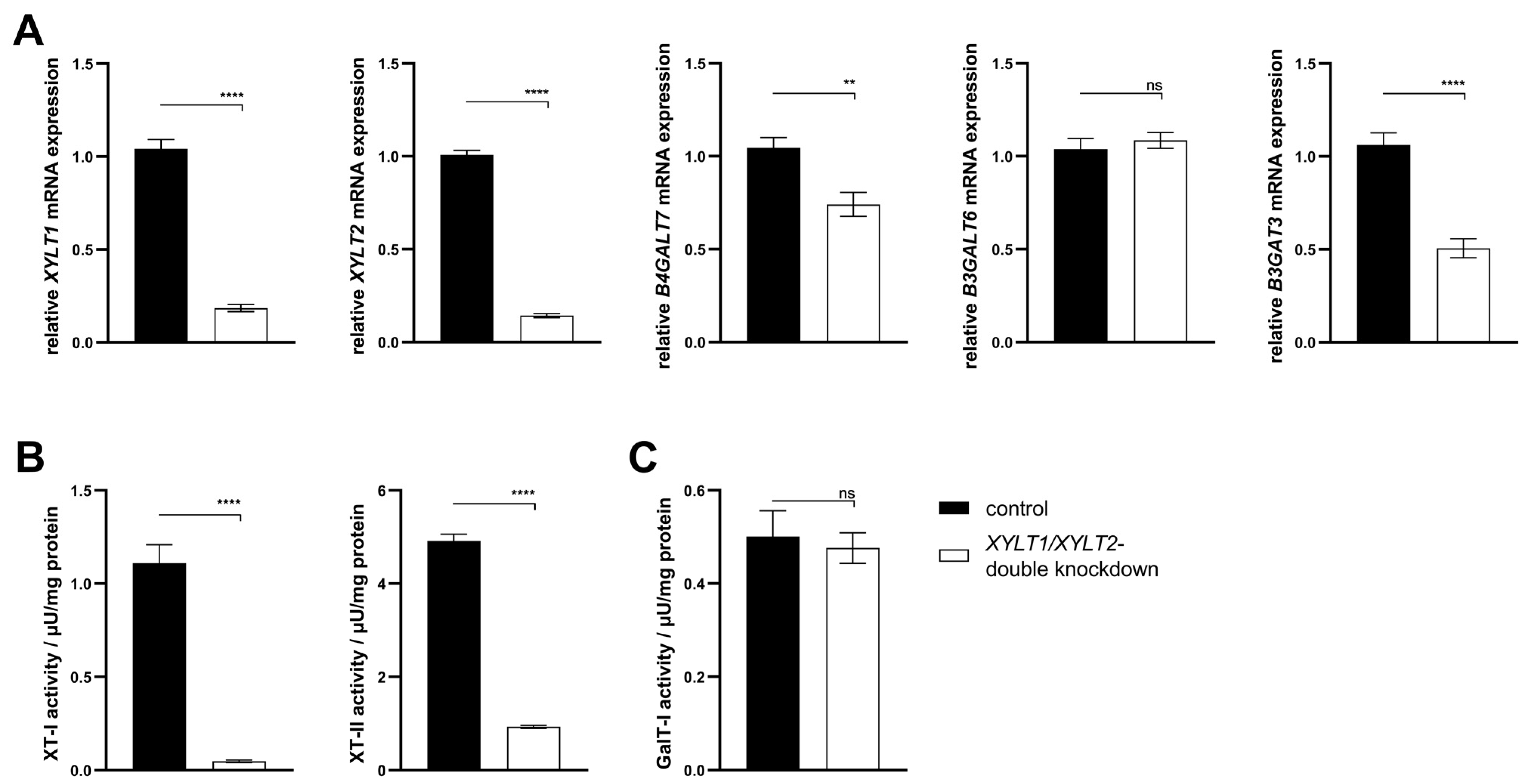
3.3. Establishment of a Simultaneous XYLT1 and XYLT2 dKD in Dermal Fibroblasts
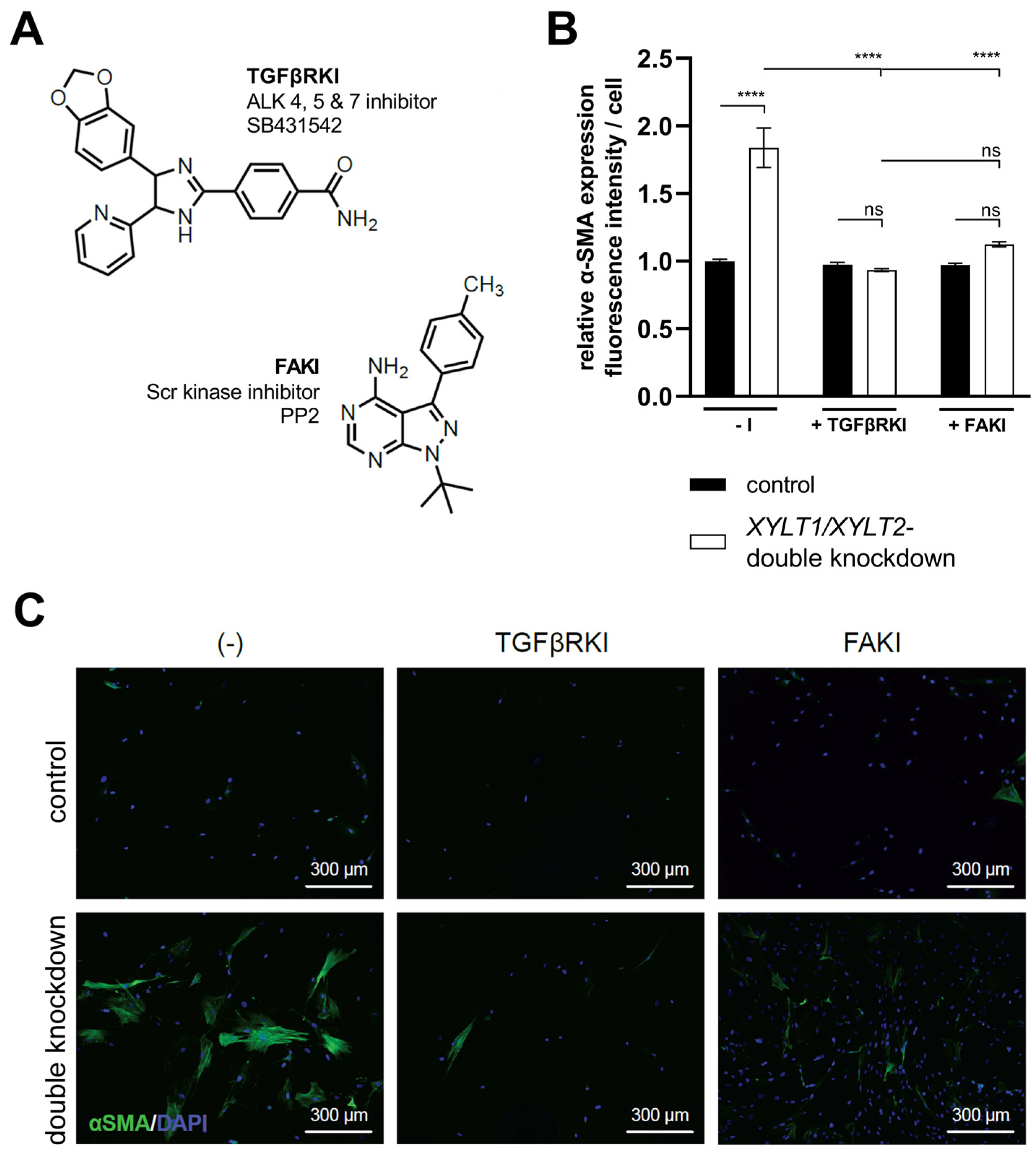
3.4. Analysis of TGFβ Signal Transduction following XYLT1/XYLT2 dKD in Dermal Fibroblasts
4. Discussion
4.1. Characterization of the Introduced Genetic Mutations
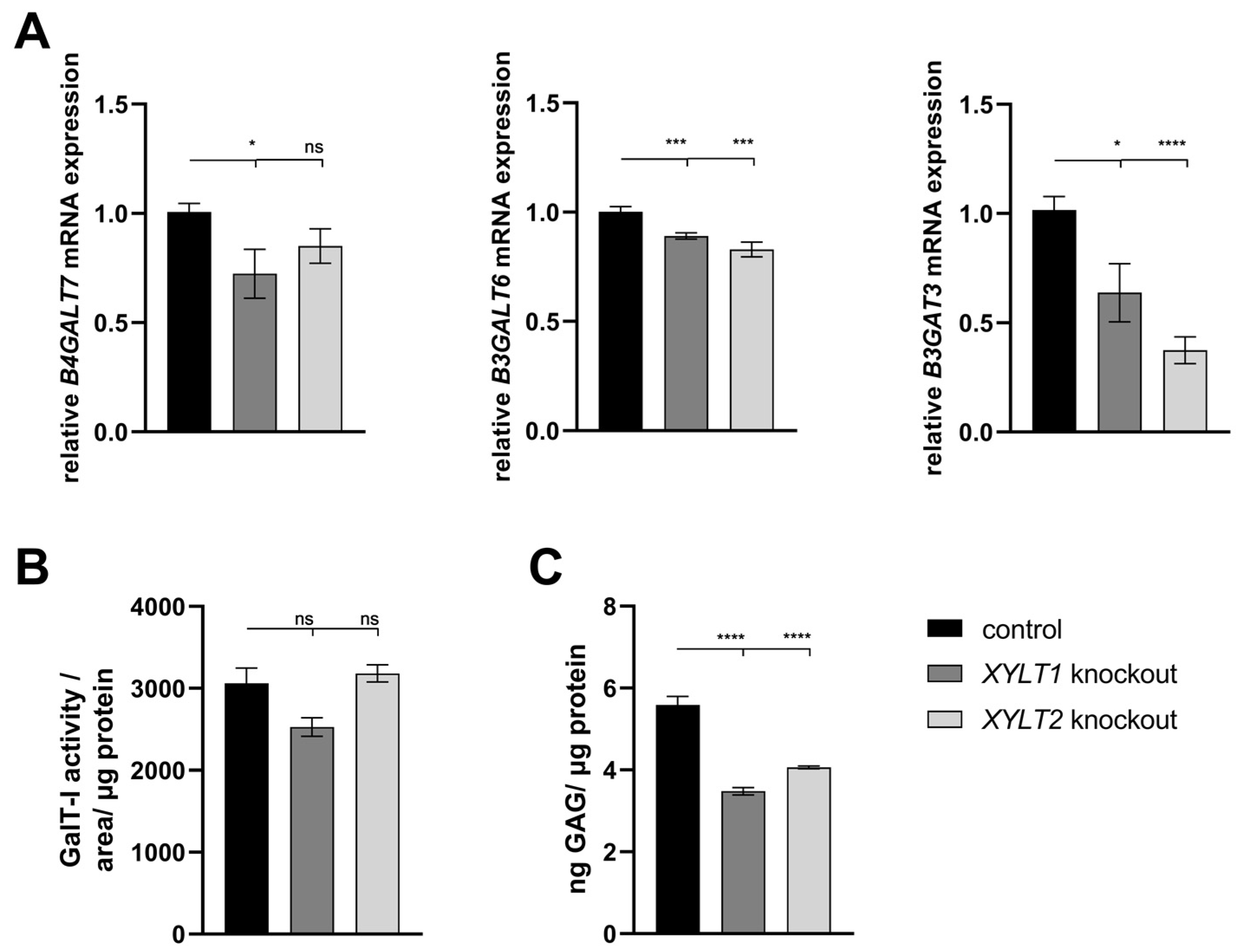
4.2. Analysis of Tetrasaccharide Linker Transferase Expressions and Their Activities
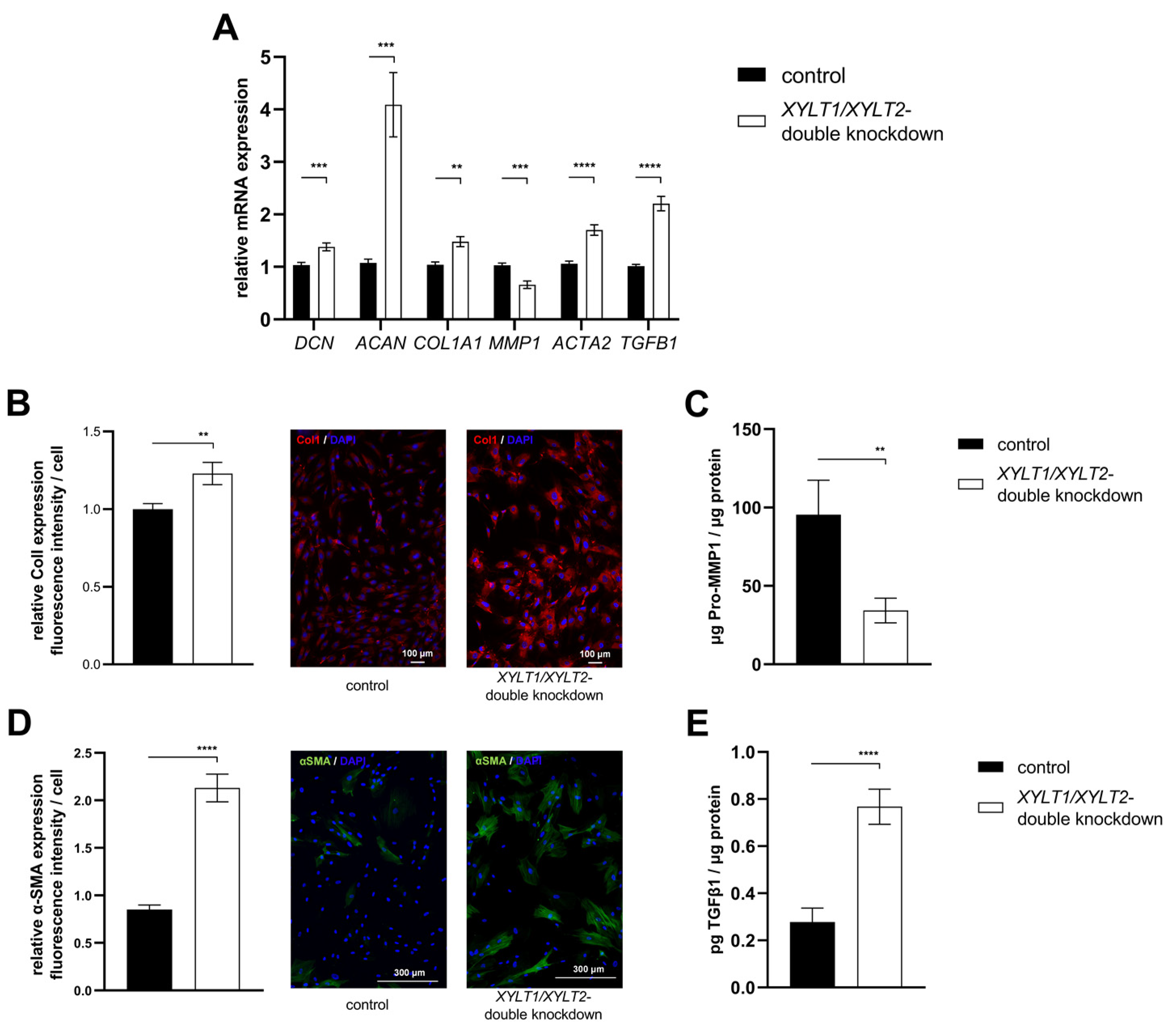
4.3. Analysis of sGAG Concentration and DCN and ACAN mRNA Expressions
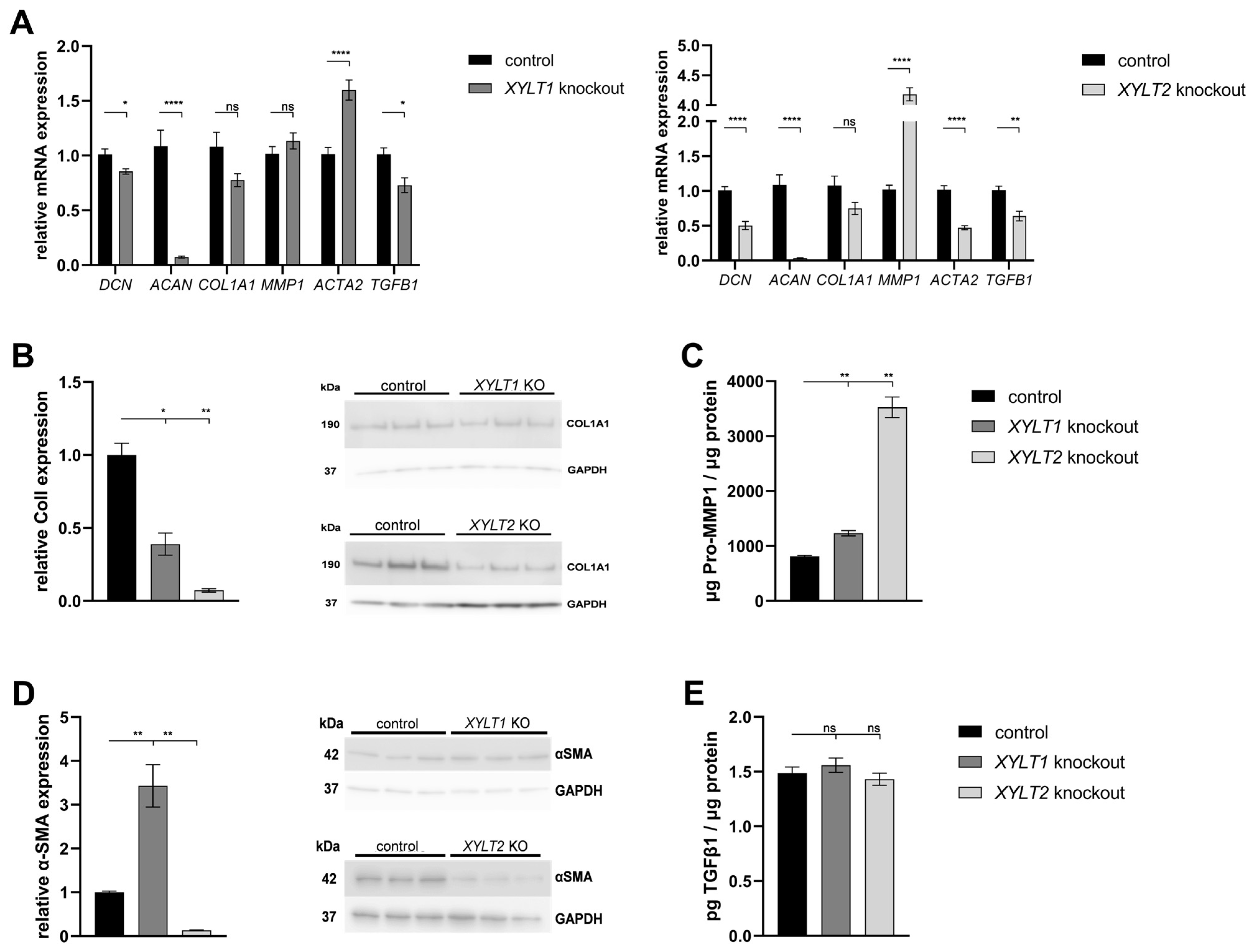
4.4. Analysis of the Expression of ECM-Associated Proteins
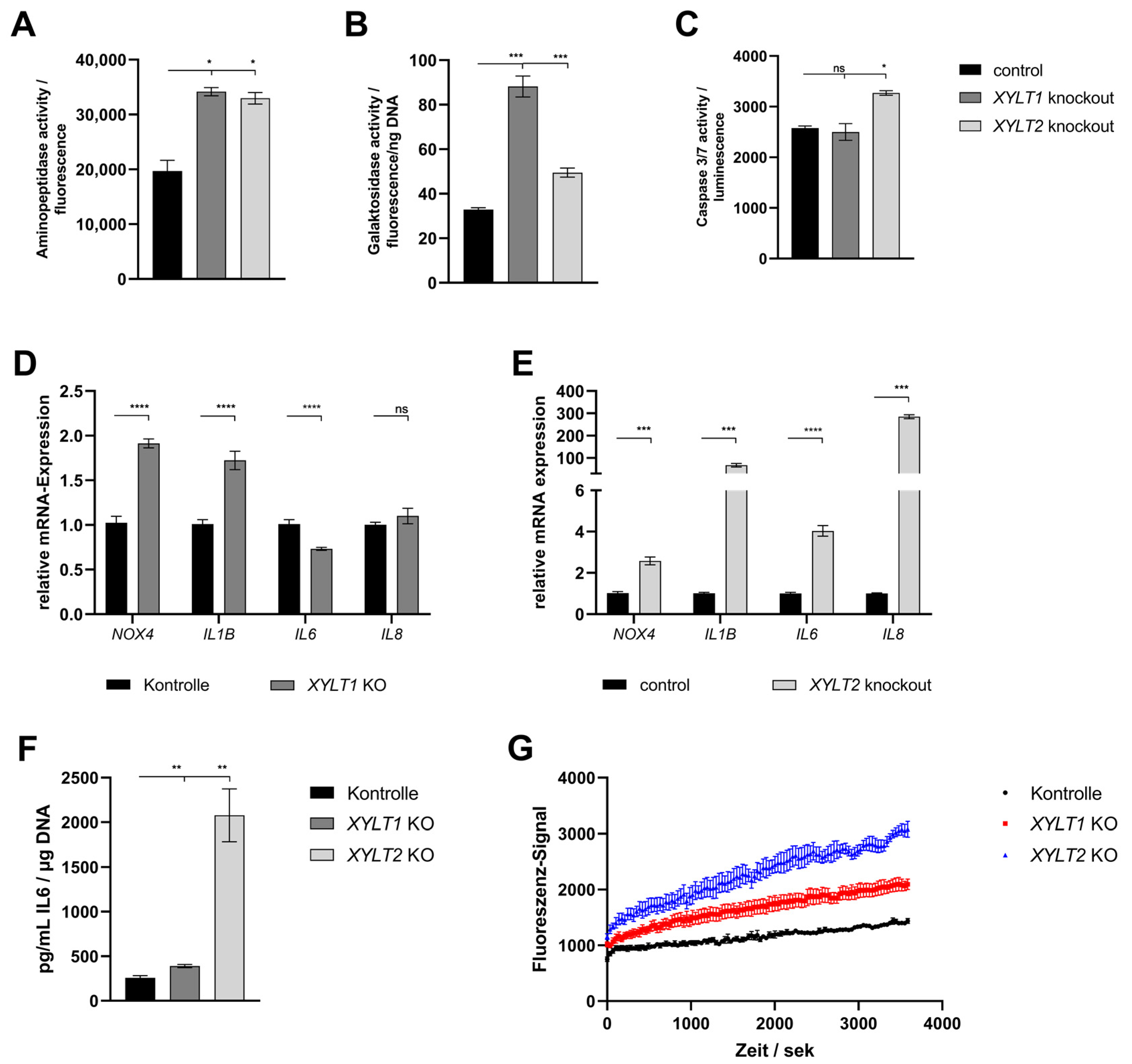
4.5. Analysis of Viability, Senescence, Apoptosis and Oxidative Stress
4.6. Characterization of XYLT1- and XYLT2-Double-Deficient Dermal Fibroblasts
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aumailley, M.; Gayraud, B. Structure and Biological Activity of the Extracellular Matrix. J. Mol. Med. 1998, 76, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Daley, W.P.; Peters, S.B.; Larsen, M. Extracellular Matrix Dynamics in Development and Regenerative Medicine. J. Cell Sci. 2008, 121, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and Interactions. Annu. Rev. Biochem. 1991, 60, 443–475. [Google Scholar] [CrossRef]
- Sugahara, K.; Kitagawa, H. Recent Advances in the Study of the Biosynthesis and Functions of Sulfated Glycosaminoglycans. Curr. Opin. Struct. Biol. 2000, 10, 518–527. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; ISBN 978-0-87969-770-9. [Google Scholar]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Zhang, H.; Feng, W.; Wang, Q.; Fan, R. Severe Phenotypes of B3GAT3-Related Disorder Caused by Two Heterozygous Variants: A Case Report and Literature Review. BMC Med. Genom. 2022, 15, 27. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S. Congenital Disorders of Deficiency in Glycosaminoglycan Biosynthesis. Front. Genet. 2021, 12, 717535. [Google Scholar] [CrossRef]
- Bui, C.; Huber, C.; Tuysuz, B.; Alanay, Y.; Bole-Feysot, C.; Leroy, J.G.; Mortier, G.; Nitschke, P.; Munnich, A.; Cormier-Daire, V. XYLT1 Mutations in Desbuquois Dysplasia Type 2. Am. J. Hum. Genet. 2014, 94, 405–414. [Google Scholar] [CrossRef]
- Silveira, C.; Leal, G.F.; Cavalcanti, D.P. Desbuquois Dysplasia Type II in a Patient with a Homozygous Mutation in XYLT1 and New Unusual Findings. Am. J. Med. Genet. Part A 2016, 170, 3043–3047. [Google Scholar] [CrossRef]
- Schreml, J.; Durmaz, B.; Cogulu, O.; Keupp, K.; Beleggia, F.; Pohl, E.; Milz, E.; Coker, M.; Ucar, S.K.; Nürnberg, G.; et al. The Missing “Link”: An Autosomal Recessive Short Stature Syndrome Caused by a Hypofunctional XYLT1 Mutation. Hum. Genet. 2014, 133, 29–39. [Google Scholar] [CrossRef] [PubMed]
- van Koningsbruggen, S.; Knoester, H.; Bakx, R.; Mook, O.; Knegt, L.; Cobben, J.M. Complete and Partial XYLT1 Deletion in a Patient with Neonatal Short Limb Skeletal Dysplasia. Am. J. Med. Genet. Part A 2016, 170, 510–514. [Google Scholar] [CrossRef]
- Jamsheer, A.; Olech, E.M.; Kozłowski, K.; Niedziela, M.; Sowińska-Seidler, A.; Obara-Moszyńska, M.; Latos-Bieleńska, A.; Karczewski, M.; Zemojtel, T. Exome Sequencing Reveals Two Novel Compound Heterozygous XYLT1 Mutations in a Polish Patient with Desbuquois Dysplasia Type 2 and Growth Hormone Deficiency. J. Hum. Genet. 2016, 61, 577–583. [Google Scholar] [CrossRef]
- Guo, L.; Elcioglu, N.H.; Iida, A.; Demirkol, Y.K.; Aras, S.; Matsumoto, N.; Nishimura, G.; Miyake, N.; Ikegawa, S. Novel and Recurrent XYLT1 Mutations in Two Turkish Families with Desbuquois Dysplasia, Type 2. J. Hum. Genet. 2017, 62, 447–451. [Google Scholar] [CrossRef]
- Al-Jezawi, N.K.; Ali, B.R.; Al-Gazali, L. Endoplasmic Reticulum Retention of Xylosyltransferase 1 (XYLT1) Mutants Underlying Desbuquois Dysplasia Type II. Am. J. Med. Genet. Part A 2017, 173, 1773–1781. [Google Scholar] [CrossRef]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef]
- Baratela, W.A.R.; Bober, M.B.; Tiller, G.E.; Okenfuss, E.; Ditro, C.; Duker, A.; Krakow, D.; Stabley, D.L.; Sol-Church, K.; Mackenzie, W.; et al. A Newly Recognized Syndrome with Characteristic Facial Features, Skeletal Dysplasia, and Developmental Delay. Am. J. Med. Genet. Part A 2012, 158, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Munns, C.F.; Fahiminiya, S.; Poudel, N.; Munteanu, M.C.; Majewski, J.; Sillence, D.O.; Metcalf, J.P.; Biggin, A.; Glorieux, F.; Fassier, F.; et al. Homozygosity for Frameshift Mutations in XYLT2 Result in a Spondylo-Ocular Syndrome with Bone Fragility, Cataracts, and Hearing Defects. Am. J. Hum. Genet. 2015, 96, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Taylan, F.; Costantini, A.; Coles, N.; Pekkinen, M.; Héon, E.; Şıklar, Z.; Berberoğlu, M.; Kämpe, A.; Kıykım, E.; Grigelioniene, G.; et al. Spondyloocular Syndrome: Novel Mutations in XYLT2 Gene and Expansion of the Phenotypic Spectrum: XYLT2 Gene Mutations in Spondyloocular Syndrome. J. Bone Miner. Res. 2016, 31, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Taylan, F.; Yavaş Abalı, Z.; Jäntti, N.; Güneş, N.; Darendeliler, F.; Baş, F.; Poyrazoğlu, Ş.; Tamçelik, N.; Tüysüz, B.; Mäkitie, O. Two Novel Mutations in XYLT2 Cause Spondyloocular Syndrome. Am. J. Med. Genet. 2017, 173, 3195–3200. [Google Scholar] [CrossRef] [PubMed]
- Umair, M.; Eckstein, G.; Rudolph, G.; Strom, T.; Graf, E.; Hendig, D.; Hoover, J.; Alanay, J.; Meitinger, T.; Schmidt, H.; et al. Homozygous XYLT2 Variants as a Cause of Spondyloocular Syndrome. Clin. Genet. 2018, 93, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Guleray, N.; Simsek Kiper, P.O.; Utine, G.E.; Boduroglu, K.; Alikasifoglu, M. Intrafamilial Variability of XYLT2-Related Spondyloocular Syndrome. Eur. J. Med. Genet. 2019, 62, 103585. [Google Scholar] [CrossRef]
- Buyukyilmaz, G.; Adiguzel, K.T.; Kılıc, E. Bisphosphonate Treatment at Spondylo-Ocular Syndrome Due to a Novel Compound Heterozygote Variant in XYLT2 and Review of the Literature. Am. J. Med Genet. Part A 2023, 191, 1581–1585. [Google Scholar] [CrossRef]
- Kausar, M.; Chew, E.G.Y.; Ullah, H.; Anees, M.; Khor, C.C.; Foo, J.N.; Makitie, O.; Siddiqi, S. A Novel Homozygous Frameshift Variant in XYLT2 Causes Spondyloocular Syndrome in a Consanguineous Pakistani Family. Front. Genet. 2019, 10, 144. [Google Scholar] [CrossRef]
- Al-Araimi, M.; Hamza, N.; Al-Hosni, A.; Al Maimani, A. Spondylo-Ocular Syndrome Due to a Novel Variant in XYLT2 in an Omani Patient. J. Pediatr. Genet. 2022, 11, 59–62. [Google Scholar] [CrossRef]
- Doddato, G.; Fabbiani, A.; Fallerini, C.; Bruttini, M.; Hadjistilianou, T.; Landi, M.; Coradeschi, C.; Grosso, S.; Tomasini, B.; Mencarelli, M.A.; et al. Corrigendum: Spondyloocular Syndrome: A Novel XYLT2 Variant with Description of the Neonatal Phenotype. Front. Genet. 2023, 14, 1143795. [Google Scholar] [CrossRef]
- Mis, E.K.; Liem, K.F.; Kong, Y.; Schwartz, N.B.; Domowicz, M.; Weatherbee, S.D. Forward Genetics Defines Xylt1 as a Key, Conserved Regulator of Early Chondrocyte Maturation and Skeletal Length. Dev. Biol. 2014, 385, 67–82. [Google Scholar] [CrossRef]
- Sivasami, P.; Poudel, N.; Munteanu, M.C.; Hudson, J.; Lovern, P.; Liu, L.; Griffin, T.; Hinsdale, M.E. Adipose Tissue Loss and Lipodystrophy in Xylosyltransferase II Deficient Mice. Int. J. Obes. 2019, 43, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Kleine, A.; Kühle, M.; Kuhn, J.; Ly, T.-D.; Schmidt, V.; Faust-Hinse, I.; Knabbe, C.; Fischer, B. A Novel SPE-UPLC-MS/MS-Based Assay for the Selective, Simultaneous Quantification of Xylosyltransferase-I and -II Activity. Biochimie 2024, 218, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Kleesiek, K.; Götting, C. Determination of Β4-Galactosyltransferase-7 Activity Using High-Performance Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry. Clin. Biochem. 2009, 42, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Ly, T.-D.; Kleine, A.; Fischer, B.; Schmidt, V.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. Identification of Putative Non-Substrate-Based XT-I Inhibitors by Natural Product Library Screening. Biomolecules 2020, 10, 1467. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.; Ohmes, J.; Ly, T.-D.; Fischer, B.; Kleine, A.; Knabbe, C.; Faust-Hinse, I. Human Xylosyltransferase I—An Important Linker between Acute Senescence and Fibrogenesis. Biomedicines 2023, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking about? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-Frequency off-Target Mutagenesis Induced by CRISPR-Cas Nucleases in Human Cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA Targeting Specificity of RNA-Guided Cas9 Nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling–from Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Plümers, R. Einfluss einer ABCC6-Defizienz auf die Remodellierung der Extrazellulären Matrix in In Vitro und Ex Vivo Gewebemodellen; Universität Bielefeld: Bielefeld, Germany, 2023. [Google Scholar]
- Fischer, B.; Schmidt, V.; Ly, T.-D.; Kleine, A.; Knabbe, C.; Faust-Hinse, I. First Characterization of Human Dermal Fibroblasts Showing a Decreased Xylosyltransferase-I Expression Induced by the CRISPR/Cas9 System. Int. J. Mol. Sci. 2022, 23, 5045. [Google Scholar] [CrossRef]
- Fischer, B. Untersuchungen zur Funktionellen Relevanz der Humanen Xylosyltransferase-Isoformen und Generierung Eines Stabilen CRISPR/Cas9-Vermittelten XYLT1 Knockouts in Dermalfibroblasten. Ph.D. Thesis, Universität Bielefel, Bielefeld, Germany, 2019. [Google Scholar]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the Linkage Region of Glycosaminoglycans: Cloning and Activity of Galactosyltransferase II, the Sixth Member of the β1,3-Galactosyltransferase Family (β3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [CrossRef]
- Taieb, M.; Ghannoum, D.; Barré, L.; Ouzzine, M. Xylosyltransferase I Mediates the Synthesis of Proteoglycans with Long Glycosaminoglycan Chains and Controls Chondrocyte Hypertrophy and Collagen Fibers Organization of in the Growth Plate. Cell Death Dis. 2023, 14, 355. [Google Scholar] [CrossRef]
- Ferencz, B.; Condac, E.; Poudel, N.; Munteanu, M.C.; Sivasami, P.; Choudhury, B.; Naidu, N.N.; Zhang, F.; Breshears, M.; Linhardt, R.J.; et al. Xylosyltransferase 2 Deficiency and Organ Homeostasis. Glycoconj. J. 2020, 37, 755–765. [Google Scholar] [CrossRef]
- Delbaere, S.; Van Damme, T.; Syx, D.; Symoens, S.; Coucke, P.; Willaert, A.; Malfait, F. Hypomorphic Zebrafish Models Mimic the Musculoskeletal Phenotype of β4GalT7-Deficient Ehlers-Danlos Syndrome. Matrix Biol. 2020, 89, 59–75. [Google Scholar] [CrossRef]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. Defective Initiation of Glycosaminoglycan Synthesis Due to B3GALT6 Mutations Causes a Pleiotropic Ehlers-Danlos-Syndrome-like Connective Tissue Disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar] [CrossRef]
- Ritelli, M.; Chiarelli, N.; Zoppi, N.; Dordoni, C.; Quinzani, S.; Traversa, M.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Insights in the Etiopathology of Galactosyltransferase II (GalT-II) Deficiency from Transcriptome-Wide Expression Profiling of Skin Fibroblasts of Two Sisters with Compound Heterozygosity for Two Novel B3GALT6 Mutations. Mol. Genet. Metab. Rep. 2015, 2, 1–15. [Google Scholar] [CrossRef]
- Delbaere, S.; De Clercq, A.; Mizumoto, S.; Noborn, F.; Bek, J.W.; Alluyn, L.; Gistelinck, C.; Syx, D.; Salmon, P.L.; Coucke, P.J.; et al. b3galt6 Knock-Out Zebrafish Recapitulate β3GalT6-Deficiency Disorders in Human and Reveal a Trisaccharide Proteoglycan Linkage Region. Front. Cell Dev. Biol. 2020, 8, 597857. [Google Scholar] [CrossRef] [PubMed]
- Baasanjav, S.; Al-Gazali, L.; Hashiguchi, T.; Mizumoto, S.; Fischer, B.; Horn, D.; Seelow, D.; Ali, B.R.; Aziz, S.A.A.; Langer, R.; et al. Faulty Initiation of Proteoglycan Synthesis Causes Cardiac and Joint Defects. Am. J. Hum. Genet. 2011, 89, 15–27. [Google Scholar] [CrossRef] [PubMed]
- von Oettingen, J.E.; Tan, W.-H.; Dauber, A. Skeletal Dysplasia, Global Developmental Delay, and Multiple Congenital Anomalies in a 5-Year-Old Boy-Report of the Second Family with B3GAT3 Mutation and Expansion of the Phenotype. Am. J. Med. Genet. A 2014, 164, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Budde, B.S.; Mizumoto, S.; Kogawa, R.; Becker, C.; Altmüller, J.; Thiele, H.; Rüschendorf, F.; Toliat, M.R.; Kaleschke, G.; Hämmerle, J.M.; et al. Skeletal Dysplasia in a Consanguineous Clan from the Island of Nias/Indonesia Is Caused by a Novel Mutation in B3GAT3. Hum. Genet. 2015, 134, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Holmborn, K.; Habicher, J.; Kasza, Z.; Eriksson, A.S.; Filipek-Gorniok, B.; Gopal, S.; Couchman, J.R.; Ahlberg, P.E.; Wiweger, M.; Spillmann, D.; et al. On the Roles and Regulation of Chondroitin Sulfate and Heparan Sulfate in Zebrafish Pharyngeal Cartilage Morphogenesis. J. Biol. Chem. 2012, 287, 33905–33916. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Yu, S.; Ye, X.; Jiang, Y.; He, P.; Shan, X. Downregulation of ACAN Is Associated with the Growth Hormone Pathway and Induces Short Stature. J. Clin. Lab. Anal. 2023, 37, e24830. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.; Chen, H.; Hassan, M.; Javed, A. Dwarfism in Homozygous Agc1CreERT Mice Is Associated with Decreased Expression of Aggrecan. Genesis 2017, 55, e23070. [Google Scholar] [CrossRef] [PubMed]
- Roughley, P.J.; Mort, J.S. The Role of Aggrecan in Normal and Osteoarthritic Cartilage. J. Exp. Orthop. 2014, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Barré, L.; Magdalou, J.; Mainard, D.; Netter, P.; Fournel-Gigleux, S.; Ouzzine, M. Modulation of Xylosyltransferase I Expression Provides a Mechanism Regulating Glycosaminoglycan Chain Synthesis during Cartilage Destruction and Repair. FASEB J. 2009, 23, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Li, Q.; Wang, C.; Patel, P.; Adams, S.M.; Doyran, B.; Nia, H.T.; Oftadeh, R.; Zhou, S.; Li, C.Y.; et al. Decorin Regulates the Aggrecan Network Integrity and Biomechanical Functions of Cartilage Extracellular Matrix. ACS Nano 2019, 13, 11320–11333. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-Allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, S.R. Matrix Metalloproteinase Interactions with Collagen and Elastin. Matrix Biol. 2015, 44–46, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Gensure, R.C.; Mäkitie, O.; Barclay, C.; Chan, C.; DePalma, S.R.; Bastepe, M.; Abuzahra, H.; Couper, R.; Mundlos, S.; Sillence, D.; et al. A Novel COL1A1 Mutation in Infantile Cortical Hyperostosis (Caffey Disease) Expands the Spectrum of Collagen-Related Disorders. J. Clin. Investig. 2005, 115, 1250–1257. [Google Scholar] [CrossRef]
- Byers, P.H.; Murray, M.L. Heritable Collagen Disorders: The Paradigm of the Ehlers—Danlos Syndrome. J. Investig. Dermatol. 2012, 132, E6–E11. [Google Scholar] [CrossRef]
- Mao, J.-R.; Bristow, J. The Ehlers-Danlos Syndrome: On beyond Collagens. J. Clin. Investig. 2001, 107, 1063–1069. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making Sense of Latent TGFβ Activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix Stiffness–Induced Myofibroblast Differentiation Is Mediated by Intrinsic Mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.W.; Li, Y.; Huang, G.; Lu, T.J.; Safwani, W.K.Z.W.; Pingguan-Murphy, B.; Xu, F. Mechanoregulation of Cardiac Myofibroblast Differentiation: Implications for Cardiac Fibrosis and Therapy. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H532–H542. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.; Nain, A.S. ECM in Differentiation: A Review of Matrix Structure, Composition and Mechanical Properties. Ann. Biomed. Eng. 2020, 48, 1071–1089. [Google Scholar] [CrossRef] [PubMed]
- Manabe, I.; Shindo, T.; Nagai, R. Gene Expression in Fibroblasts and Fibrosis: Involvement in Cardiac Hypertrophy. Circ. Res. 2002, 91, 1103–1113. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Horikawa, I.; Harris, C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet. Pathol. 2020, 57, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells In Vitro and In Vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Meyer, P.; Maity, P.; Burkovski, A.; Schwab, J.; Müssel, C.; Singh, K.; Ferreira, F.F.; Krug, L.; Maier, H.J.; Wlaschek, M.; et al. A Model of the Onset of the Senescence Associated Secretory Phenotype after DNA Damage Induced Senescence. PLOS Comput. Biol. 2017, 13, e1005741. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Chronic Inflammation and Oxidative Stress in the Genesis and Perpetuation of Cancer: Role of Lipid Peroxidation, DNA Damage, and Repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Mochitate, K.; Pawelek, P.; Grinnell, F. Stress Relaxation of Contracted Collagen Gels: Disruption of Actin Filament Bundles, Release of Cell Surface Fibronectin, and down-Regulation of DNA and Protein Synthesis. Exp. Cell Res. 1991, 193, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-M.; Xu, S.-Y.; Feng, Y.-Z.; Cheng, Y.-R.; Xiong, J.-B.; Zhou, Y.; Guan, C.-X. The Role of NOX4 in Pulmonary Diseases. J. Cell. Physiol. 2021, 236, 1628–1637. [Google Scholar] [CrossRef]
- McCawley, L.J.; Matrisian, L.M. Matrix Metalloproteinases: They’re Not Just for Matrix Anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Papadopoulou, A.; Fotopoulou, A.; Tsimelis, S.; Bassiony, H.; Yiacoumettis, A.M.; Panagiotou, P.N.; Pratsinis, H.; Kletsas, D. Down-Regulation of the Proteoglycan Decorin Fills in the Tumor-Promoting Phenotype of Ionizing Radiation-Induced Senescent Human Breast Stromal Fibroblasts. Cancers 2021, 13, 1987. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Li, G.; Huang, Y.; Fu, Z.; Song, X.; Chen, C.; Yang, L. Synergistically Regulated Spontaneous Calcium Signaling Is Attributed to Cartilaginous Extracellular Matrix Metabolism. J. Cell. Physiol. 2019, 234, 9711–9722. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Hattori, A.; Ishii, Y.; Mizutani, S.; Tsujimoto, M. Enzymatic Properties of Human Aminopeptidase A: Regulation of Its Enzymatic Activity by Calcium and Angiotensin IV. J. Biol. Chem. 2006, 281, 23503–23513. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, X.; Gao, Z.Y.; Liu, K.; Hou, Y.; Zheng, J. The Role of miR-320a and IL-1β in Human Chondrocyte Degradation. Bone Jt. Res. 2017, 6, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.-P. Senescence-Associated IL-6 and IL-8 Cytokines Induce a Self- and Cross-Reinforced Senescence/Inflammatory Milieu Strengthening Tumorigenic Capabilities in the MCF-7 Breast Cancer Cell Line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef]
- Khan, K.; Xu, S.; Nihtyanova, S.; Derrett-Smith, E.; Abraham, D.; Denton, C.P.; Ong, V.H. Clinical and Pathological Significance of Interleukin 6 Overexpression in Systemic Sclerosis. Ann. Rheum. Dis. 2012, 71, 1235–1242. [Google Scholar] [CrossRef]
- Kenny, F.N.; Marcotti, S.; De Freitas, D.B.; Drudi, E.M.; Leech, V.; Bell, R.E.; Easton, J.; Díaz-de-la-Loza, M.-D.-C.; Fleck, R.; Allison, L.; et al. Autocrine IL-6 Drives Cell and Extracellular Matrix Anisotropy in Scar Fibroblasts. Matrix Biol. 2023, 123, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Ju, X.; Sun, H.; Finnerty, C.C.; Herndon, D.N.; Brasier, A.R. The IL-6 Trans-Signaling-STAT3 Pathway Mediates ECM and Cellular Proliferation in Fibroblasts from Hypertrophic Scar. J. Investig. Dermatol. 2013, 133, 1212–1220. [Google Scholar] [CrossRef]
- Fischer, B.; Ly, T.-D.; Schmidt, V.; Hendig, D.; Kuhn, J.; Knabbe, C.; Faust, I. Xylosyltransferase-Deficient Human HEK293 cells Show a Strongly Reduced Proliferation Capacity and Viability. Biochem. Biophys. Res. Commun. 2020, 521, 507–513. [Google Scholar] [CrossRef]
- Seidler, D.G.; Faiyaz-Ul-Haque, M.; Hansen, U.; Yip, G.W.; Zaidi, S.H.E.; Teebi, A.S.; Kiesel, L.; Götte, M. Defective Glycosylation of Decorin and Biglycan, Altered Collagen Structure, and Abnormal Phenotype of the Skin Fibroblasts of an Ehlers-Danlos Syndrome Patient Carrying the Novel Arg270Cys Substitution in Galactosyltransferase I (β4GalT-7). J. Mol. Med. 2006, 84, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, D.; Saldeen, T.; Mehta, J.L. TGF-β1 Attenuates Myocardial Ischemia-Reperfusion Injury via Inhibition of Upregulation of MMP-1. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1612–H1617. [Google Scholar] [CrossRef]
- Siani, A.; Tirelli, N. Myofibroblast Differentiation: Main Features, Biomedical Relevance, and the Role of Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 21, 768–785. [Google Scholar] [CrossRef]
- Faust, I.; Roch, C.; Kuhn, J.; Prante, C.; Knabbe, C.; Hendig, D. Human Xylosyltransferase-I—A New Marker for Myofibroblast Differentiation in Skin Fibrosis. Biochem. Biophys. Res. Commun. 2013, 436, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Melchior-Becker, A.; Dai, G.; Ding, Z.; Schäfer, L.; Schrader, J.; Young, M.F.; Fischer, J.W. Deficiency of Biglycan Causes Cardiac Fibroblasts to Differentiate into a Myofibroblast Phenotype. J. Biol. Chem. 2011, 286, 17365–17375. [Google Scholar] [CrossRef]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix Control of Transforming Growth Factor-Function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.S.; Bernstein, A.M.; Benezra, M.; Gelman, I.H.; Taliana, L.; Masur, S.K. FAK-Dependent Regulation of Myofibroblast Differentiation. FASEB J. 2006, 20, 1006–1008. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | 5′–3′ Sequence | IDT Label |
|---|---|---|
| XYLT1 | CTTACTGCCGCCACAAGTTA | XYLT1.1.AA |
| XYLT2 | GGTTACTGCCCGTACCACCT | XYLT2.1.AB |
| Target | 5′–3′ Sequence |
|---|---|
| XYLT1 | GCAUCAUGCUACCAAUCUGtt ttCGUAGUACGAUGGUUAGAC |
| XYLT2 | GGCCGUUUAUCACGAGCAGtt ttCCGGCAAUAGUGCUCGUC |
| Target | Sequence | TA/°C | Product Size/bp |
|---|---|---|---|
| ACAN | CACCCCATGCAATTTGAG GCCACTGTGCCCTTTTTA | 63 | 158 |
| ACTA2 | GACCGAATGCAGAAGGAG CGGTGGACAATGGAAGG | 59 | 169 |
| B2M | TGTGCTCGCGCTACTCTCTCTT CGGATGGATGAAACCCAGACA | 63 | 137 |
| B3GALT6 | CCCCGCTGTGGTCTTTGTTG CGCCCCCGTTTCTTCCTC | 63 | 188 |
| B3GAT3 | GCTGTTTGAGGAGATGCGCTG TCAGAAGACTGCTCTCCAGGT | 63 | 251 |
| B4GALT7 | GCCATGCACAGTGATCAGAG CCCTACACTGTGTCTCTGCA | 63 | 196 |
| COL1A1 | GATGTGCCACTCTGACT GGGTTCTTGCTGATG | 63 | 151 |
| DCN | CCTTCCGCTGTCAATG GCAGGTCTAGCAGAGTTG | 63 | 102 |
| GAPDH | AGGTCGGAGTCAACGGAT TCCTGGAAGATGGTGATG | 59 | 223 |
| IL1B | ACAGATGAAGTGCTCCTTCCA GTCGGAGATTCGTAGCTGGAT | 63 | 73 |
| IL6 | ACAGCCACTCACCTCTTCAG GTGCCTCTTTGCTGCTTTCAC | 63 | 122 |
| IL8 | GAACTGAGAGTGATTGAGAGTGGA CTCTTCAAAAACTTCTCCACAACC | 63 | 125 |
| MMP1 | AGAAACACAAGAGCAAGATGTG TGGCGTGTAATTTTCAATCCTGT | 63 | 298 |
| NOX4 | CTTCCGTTGGTTTGCAGATT GAATTGGGTCCACAACAGA | 63 | 246 |
| RPL13A | CGGAAGGTGGTGGTCGTA CTCGGGAAGGGTTGGTGT | 63 | 115 |
| SDHA | AACTCGCTCTTGGACCTG GAGTCGCAGTTCCGATGT | 63 | 177 |
| TGFB1 | GCGATACCTCAGCAACC ACGCAGCAGTTCTTCTCC | 63 | 331 |
| XYLT1 | GAAGCCGTGGTGAATCAG CGGTCAGCAAGGAAGTAG | 63 | 281 |
| XYLT2 | ACACAGATGACCCGCTTGTGG TTGGTGACCCGCAGGTTGTTG | 63 | 139 |
| Primary Antibody | Dilution | Secondary Antibody | Dilution |
|---|---|---|---|
| Rabbit-anti-COL1A1 (ab34710; Abcam, Cambridge, UK) | 1:10,000 | Goat-anti-rabbit IgG-HRP polyclonal (ab150113; Abcam, Cambridge, UK) | 1:5000 |
| Mouse-anti-αSMA (GA61161-2; Cell Signaling Technology, Cambridge, UK) | 1:1000 | Horse-anti-mouse igG-HRP polyclonal (ab150078; Abcam, Cambridge, UK) | 1:2000 |
| Mouse-anti-gapdh (ab8245; Abcam, Cambridge, UK) | 1:5000 | Horse-anti-mouse igG-HRP polyclonal (ab150078; Abcam, Cambridge, UK) | 1:2000 |
| Primary Antibody | Dilution | Secondary Antibody | Dilution |
|---|---|---|---|
| Rabbit-anti-COL1A1 (ab34710; Abcam, Cambridge, UK) | 1:100 | Goat-anti-rabbit (Alexa-555) (A-32732; Thermo Fisher Scientific, USA) | 1:200 |
| Mouse-anti-αSMA (GA61161-2; Cell Signaling Technology, Cambridge, UK) | 1:50 | Goat-anti-mouse (Alexa-488) (A-11001; Thermo Fisher Scientific, USA) | 1:200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleine, A.; Kühle, M.; Ly, T.-D.; Schmidt, V.; Faust-Hinse, I.; Knabbe, C.; Fischer, B. Xylosyltransferase-Deficiency in Human Dermal Fibroblasts Induces Compensatory Myofibroblast Differentiation and Long-Term ECM Reduction. Biomedicines 2024, 12, 572. https://doi.org/10.3390/biomedicines12030572
Kleine A, Kühle M, Ly T-D, Schmidt V, Faust-Hinse I, Knabbe C, Fischer B. Xylosyltransferase-Deficiency in Human Dermal Fibroblasts Induces Compensatory Myofibroblast Differentiation and Long-Term ECM Reduction. Biomedicines. 2024; 12(3):572. https://doi.org/10.3390/biomedicines12030572
Chicago/Turabian StyleKleine, Anika, Matthias Kühle, Thanh-Diep Ly, Vanessa Schmidt, Isabel Faust-Hinse, Cornelius Knabbe, and Bastian Fischer. 2024. "Xylosyltransferase-Deficiency in Human Dermal Fibroblasts Induces Compensatory Myofibroblast Differentiation and Long-Term ECM Reduction" Biomedicines 12, no. 3: 572. https://doi.org/10.3390/biomedicines12030572
APA StyleKleine, A., Kühle, M., Ly, T.-D., Schmidt, V., Faust-Hinse, I., Knabbe, C., & Fischer, B. (2024). Xylosyltransferase-Deficiency in Human Dermal Fibroblasts Induces Compensatory Myofibroblast Differentiation and Long-Term ECM Reduction. Biomedicines, 12(3), 572. https://doi.org/10.3390/biomedicines12030572