
Comprehensive Transcriptomic Profiling of Diverse Brain Tumor Types Uncovers Complex Structures of the Brain Tumor Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Datasets
2.2. Microarray Data Processing and Immune Signature Expression Analysis
2.3. Single-Cell RNA Sequencing Data Processing
2.4. Consensus Clustering Using Immune-Related Genes
2.5. Gene Set Variation Analysis (GSVA) and Gene Set Enrichment Analysis (GSEA)
2.6. Differentially Expressed Gene (DEG) Analysis with Gene Ontology (GO) Enrichment
2.7. Visualization and Statistical Analysis
3. Results
3.1. Landscape of the Immune Signature Expression in the TME of Brain Tumors
3.1.1. Depiction of the Immune Signature Expression in Overall Brain Tumors
3.1.2. Comparison of Immune Expression across Brain Tumor Types
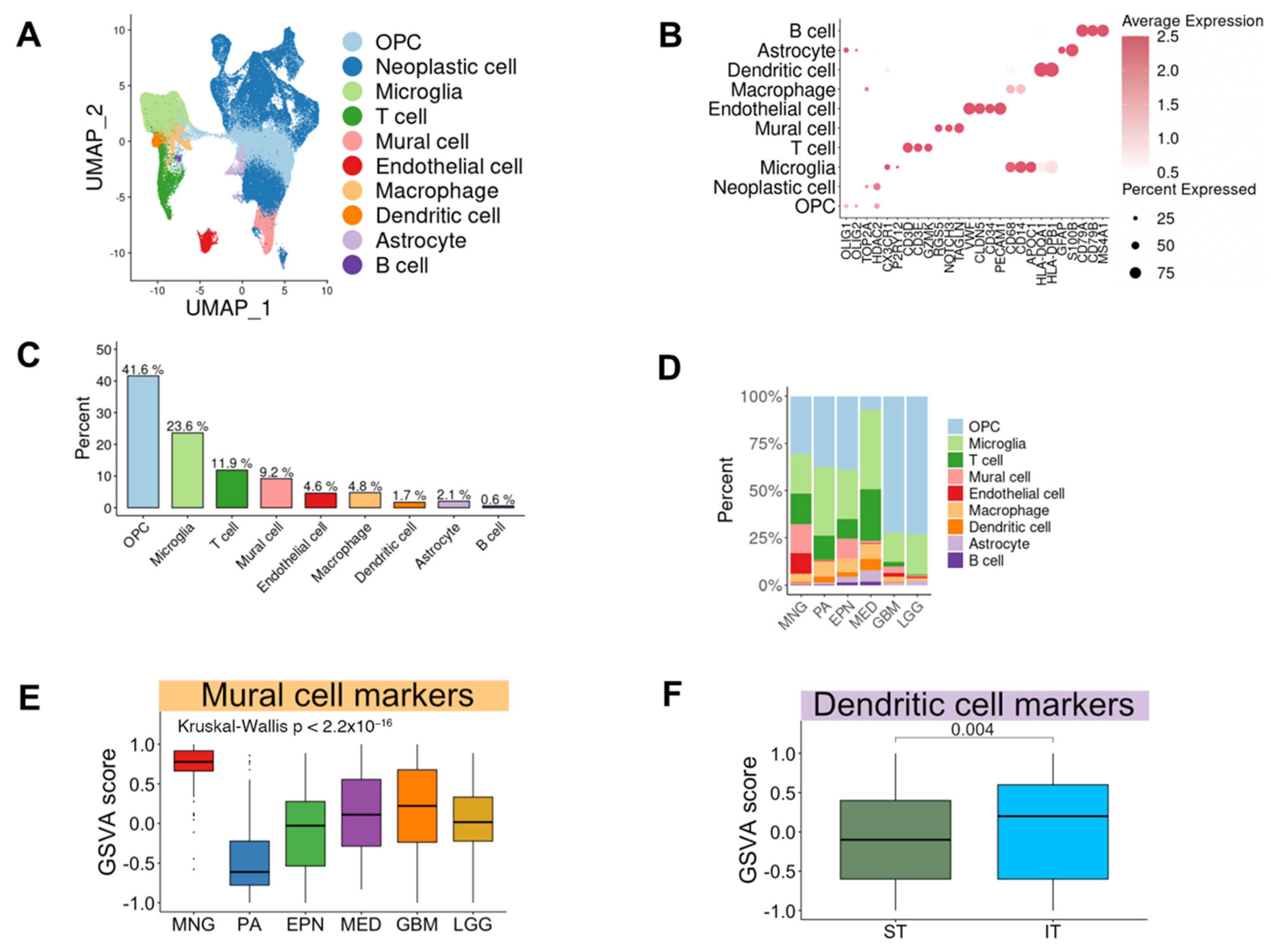
3.2. Single-Cell RNA Sequencing Revealed Cellular Heterogeneity in Various Brain Tumors
3.2.1. The Composition of Cell Types in Brain Tumors
3.2.2. Difference of Cell Type Composition across Brain Tumor Types
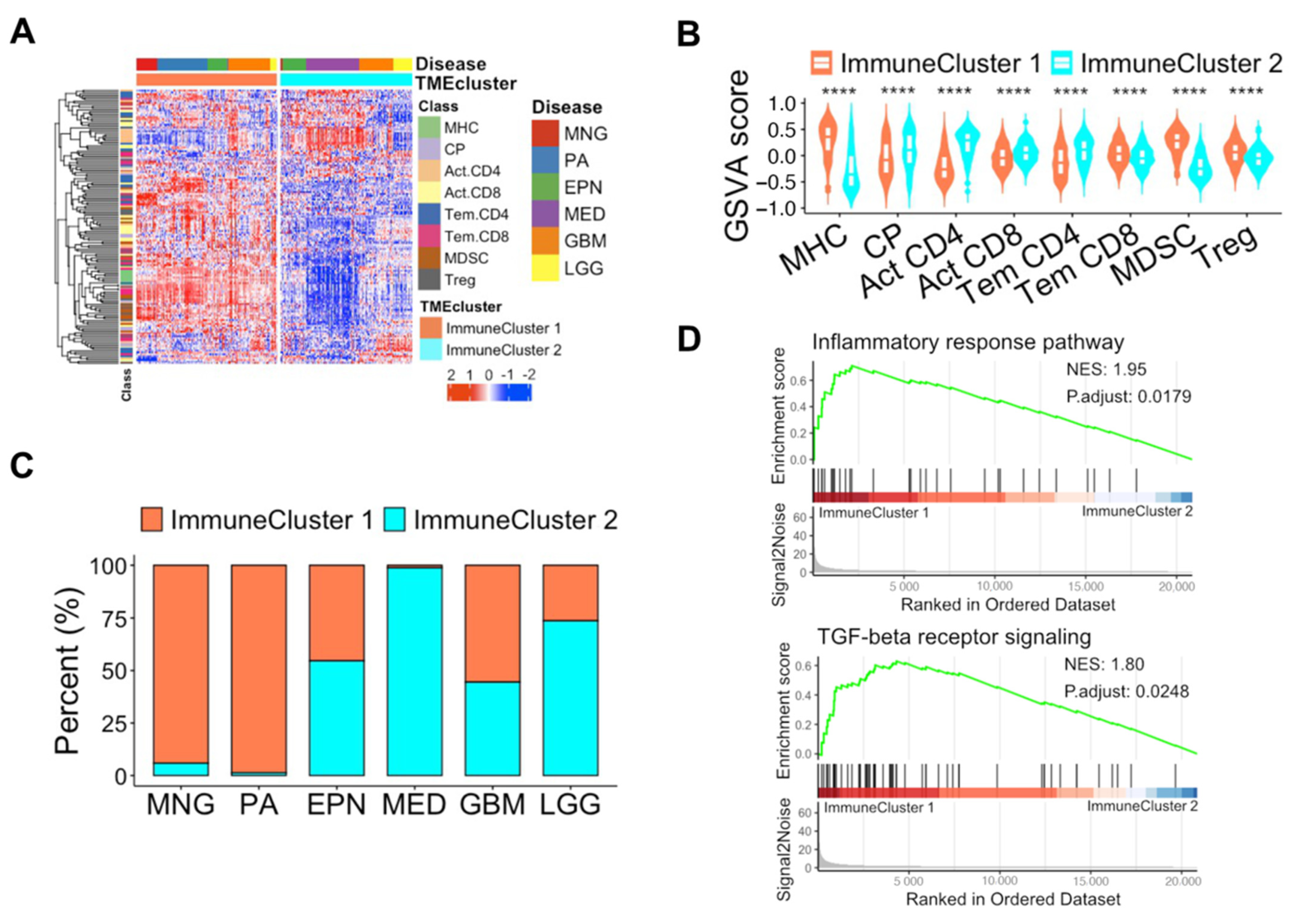
3.3. Unique Features of Immune Subpopulations Based on Unsupervised Learning
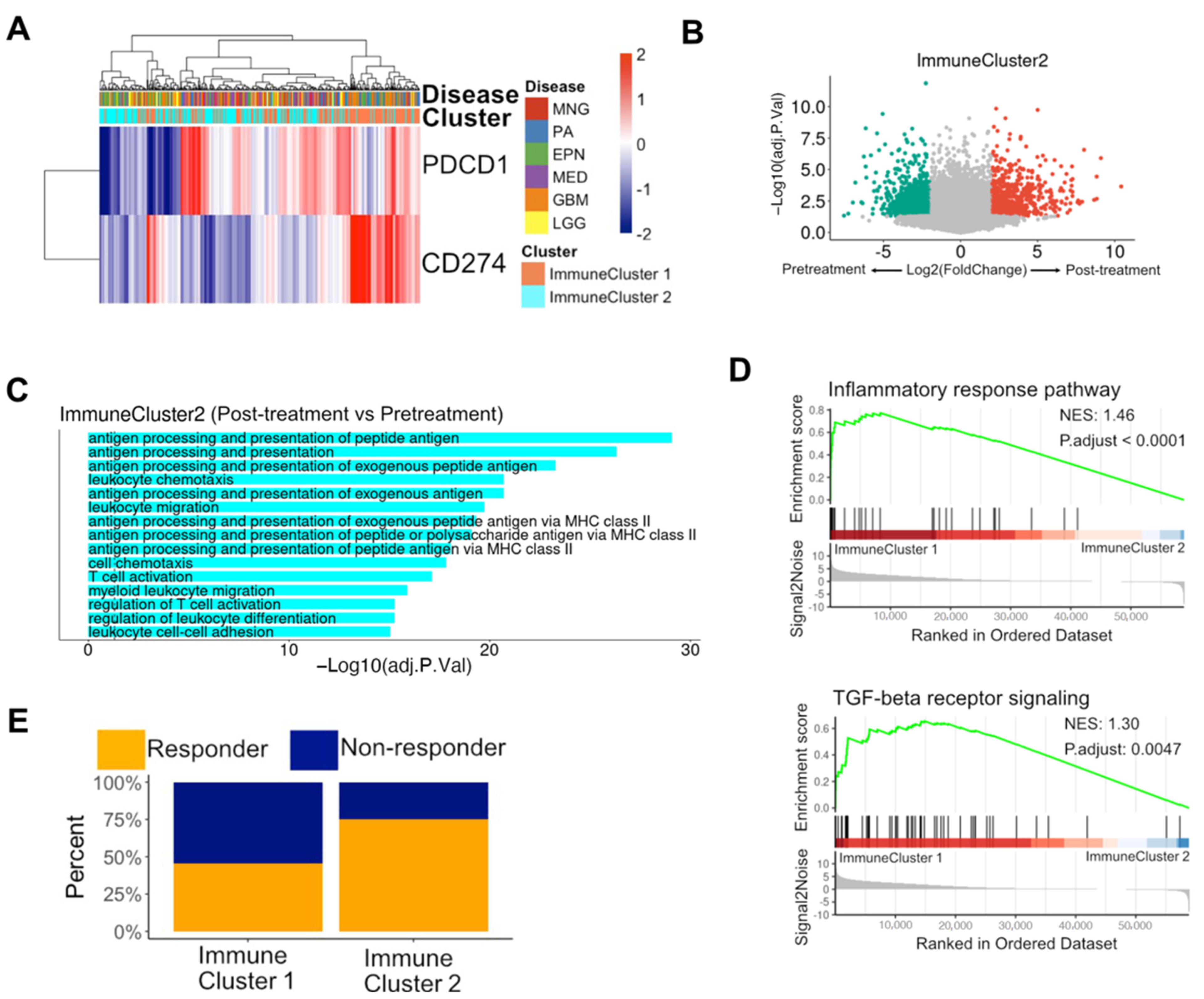
3.4. Immune Subpopulations were Potential Biomarkers of Reaction to PD-1 Blockade in Glioblastomas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McKinney, P.A. Brain tumours: Incidence, survival, and aetiology. J. Neurol. Neurosurg. Psychiatry 2004, 75, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Vera-Bolanos, E.; Aldape, K.; Yuan, Y.; Wu, J.; Wani, K.; Necesito-Reyes, M.J.; Colman, H.; Dhall, G.; Lieberman, F.S.; Metellus, P.; et al. Clinical course and progression-free survival of adult intracranial and spinal ependymoma patients. Neuro-Oncology 2015, 17, 440–447. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Focusing on brain tumours and brain metastasis. Nat. Rev. Cancer 2020, 20, 1. [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Levy, C.; Allouache, D.; Lacroix, J.; Dugue, A.E.; Supiot, S.; Campone, M.; Mahe, M.; Kichou, S.; Leheurteur, M.; Hanzen, C.; et al. REBECA: A phase I study of bevacizumab and whole-brain radiation therapy for the treatment of brain metastasis from solid tumours. Ann. Oncol. 2014, 25, 2351–2356. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Venur, V.A.; Preusser, M.; Ahluwalia, M.S. Immune Checkpoint Inhibitors in Brain Metastases: From Biology to Treatment. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e116–e122. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Ricard, D.; Idbaih, A.; Ducray, F.; Lahutte, M.; Hoang-Xuan, K.; Delattre, J.Y. Primary brain tumours in adults. Lancet 2012, 379, 1984–1996. [Google Scholar] [CrossRef]
- Lyon, J.G.; Mokarram, N.; Saxena, T.; Carroll, S.L.; Bellamkonda, R.V. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32. [Google Scholar] [CrossRef]
- Caccese, M.; Indraccolo, S.; Zagonel, V.; Lombardi, G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit. Rev. Oncol. Hemat. 2019, 135, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.R.; Kong, Z.R.; Ma, W.B. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vacc. Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Faisal, S.M.; Castro, M.G.; Lowenstein, P.R. Combined cytotoxic and immune-stimulatory gene therapy using Ad-TK and Ad-Flt3L: Translational developments from rodents to glioma patients. Mol. Ther. 2023, 31, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Umemura, Y.; Orringer, D.; Junck, L.; Varela, M.L.; West, M.E.J.; Faisal, S.M.; Comba, A.; Heth, J.; Sagher, O.; Leung, D.; et al. Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: A phase 1, first-in- human trial. Lancet Oncol. 2023, 24, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Rainov, N.G.; Grp, G.I.S. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Liu, J.; Patel, S.; Cloughesy, T.; Lai, A.; Farooqi, H.; Seligson, D.; Dong, J.; Liau, L.; Becker, D.; et al. Genomic Landscape of Meningiomas. Brain Pathol. 2010, 20, 751–762. [Google Scholar] [CrossRef]
- Sharma, M.K.; Mansur, D.B.; Reifenberger, G.; Perry, A.; Leonard, J.R.; Aldape, K.D.; Albin, M.G.; Emnett, R.J.; Loeser, S.; Watson, M.A.; et al. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res. 2007, 67, 890–900. [Google Scholar] [CrossRef]
- Zakrzewski, K.; Jarzab, M.; Pfeifer, A.; Oczko-Wojciechowska, G.; Jarzab, B.; Liberski, P.P.; Zakrzewska, M. Transcriptional profiles of pilocytic astrocytoma are related to their three different locations, but not to radiological tumor features. BMC Cancer 2015, 15, 778. [Google Scholar] [CrossRef] [PubMed]
- Donson, A.M.; Birks, D.; Barton, V.N.; Kleinschmidt-DeMasters, B.K.; Handler, M.H.; Waziri, A.E.; Foreman, N.K. Immune Gene and Cell Enrichment Is Associated with a Good Prognosis in Ependymoma. Neuro-Oncology 2009, 183, 7428–7440. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Donson, A.M.; Nakachi, I.; Griesinger, A.M.; Birks, D.K.; Amani, V.; Hemenway, M.S.; Liu, A.K.; Wang, M.; Hankinson, T.C.; et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014, 127, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Schouten-van Meeteren, N.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.; Jones, D.T.W.; Korshunov, A.; Tonjes, M.; Plass, C.; Jabado, N.; Pfister, S.M. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Neuro-Oncology 2012, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H.; Gietzelt, J.; Hentschel, B.; Westphal, M.; Simon, M.; et al. Molecular characterization of long-term survivors of glioblastoma using genome-and transcriptome-wide profiling. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef]
- Sun, L.X.; Hui, A.M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef]
- Lambert, S.R.; Witt, H.; Hovestadt, V.; Zucknick, M.; Kool, M.; Pearson, D.M.; Korshunov, A.; Ryzhova, M.; Ichimura, K.; Jabado, N.; et al. Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathol. 2013, 126, 291–301. [Google Scholar] [CrossRef]
- Griesinger, A.M.; Birks, D.K.; Donson, A.M.; Amani, V.; Hoffman, L.M.; Waziri, A.; Wang, M.; Handler, M.H.; Foreman, N.K. Characterization of Distinct Immunophenotypes across Pediatric Brain Tumor Types. J. Immunol. 2013, 191, 4880–4888. [Google Scholar] [CrossRef]
- Sean, D.; Meltzer, P.S. GEOquery: A bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar]
- Vasudevan, H.N.; Choudhury, A.; Hilz, S.; Villanueva-Meyer, J.E.; Chen, W.C.; Lucas, C.H.G.; Braunstein, S.E.; Bush, N.A.O.; Butowski, N.; Pekmezci, M.; et al. Intratumor and informatic heterogeneity influence meningioma molecular classification. Acta Neuropathol. 2022, 144, 579–583. [Google Scholar] [CrossRef]
- Gillen, A.E.; Riemondy, K.A.; Amani, V.; Griesinger, A.M.; Gilani, A.; Venkataraman, S.; Madhavan, K.; Prince, E.; Sanford, B.; Hankinson, T.C.; et al. Single-Cell RNA Sequencing of Childhood Ependymoma Reveals Neoplastic Cell Subpopulations That Impact Molecular Classification and Etiology. Cell Rep. 2020, 32, 108023. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Venkataraman, S.; Willard, N.; Nellan, A.; Sanford, B.; Griesinger, A.M.; Amani, V.; Mitra, S.; Hankinson, T.C.; Handler, M.H.; et al. Neoplastic and immune single-cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro-Oncology 2022, 24, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; He, L.Q.; Lugano, R.; Zhang, Y.Y.; Cao, H.Y.; He, Q.Y.; Chao, M.; Liu, B.X.; Cao, Q.Z.; Wang, J.H.; et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021, 6, e150861. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Paolella, B.R.; Bergthold, G.; Pelton, K.; Becker, S.; Jones, R.; Sinai, C.E.; Malkin, H.; Huang, Y.; Grimmet, L.; et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. Commun. 2019, 10, 3731. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.C.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.F.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Hazra, A.; Gogtay, N. Biostatistics Series Module 3: Comparing Groups: Numerical Variables. Indian J. Dermatol. 2016, 61, 251–260. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cistaro, A.; Albano, D.; Alongi, P.; Laudicella, R.; Pizzuto, D.A.; Formica, G.; Romagnolo, C.; Stracuzzi, F.; Frantellizzi, V.; Piccardo, A.; et al. The Role of PET in Supratentorial and Infratentorial Pediatric Brain Tumors. Curr. Oncol. 2021, 28, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Chen, W.; Zhu, Y.; Zhang, P. Clinical features associated with the efficacy of chemotherapy in patients with glioblastoma (GBM): A surveillance, epidemiology, and end results (SEER) analysis. BMC Cancer 2021, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Tubo, N.J.; Jenkins, M.K. CD4+ T Cells: Guardians of the phagosome. Clin. Microbiol. Rev. 2014, 27, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.J.; Lesniak, M.S.; James, C.D.; Heimberger, A.B.; Chen, P.W. Context-Dependent Glioblastoma?Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol. 2021, 42, 280–292. [Google Scholar] [CrossRef]
- Gastfriend, B.D.; Foreman, K.L.; Katt, M.E.; Palecek, S.P.; Shusta, E.V. Integrative analysis of the human brain mural cell transcriptome. J. Cereb. Blood Flow Metab. 2021, 41, 3052–3068. [Google Scholar] [CrossRef]
- Wang, A.Z.; Bowman-Kirigin, J.A.; Desai, R.; Kang, L.I.; Patel, P.R.; Patel, B.; Khan, S.M.; Bender, D.; Marlin, M.C.; Liu, J.X.; et al. Single-cell profiling of human dura and meningioma reveals cellular meningeal landscape and insights into meningioma immune response. Genome Med. 2022, 14, 49. [Google Scholar] [CrossRef]
- Ostman, A.; Corvigno, S. Microvascular Mural Cells in Cancer. Trends Cancer 2018, 4, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Immune Heterogeneity in Neuroinflammation: Dendritic Cells in the Brain. J. Neuroimmune Pharm. 2013, 8, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Y.; Dai, Y.; Cheng, J.N.; Gong, Z.; Feng, Y.; Sun, C.; Jia, Q.; Zhu, B. Immune Landscape of Colorectal Cancer Tumor Microenvironment from Different Primary Tumor Location. Front. Immunol. 2018, 9, 1578. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Di Nunno, V.; Aprile, M.; Gatto, L.; Tosoni, A.; Ranieri, L.; Bartolini, S.; Franceschi, E. Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab? Cancers 2023, 15, 1042. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Cho, H.J. Comprehensive Transcriptomic Profiling of Diverse Brain Tumor Types Uncovers Complex Structures of the Brain Tumor Microenvironment. Biomedicines 2024, 12, 506. https://doi.org/10.3390/biomedicines12030506
Choi J, Cho HJ. Comprehensive Transcriptomic Profiling of Diverse Brain Tumor Types Uncovers Complex Structures of the Brain Tumor Microenvironment. Biomedicines. 2024; 12(3):506. https://doi.org/10.3390/biomedicines12030506
Chicago/Turabian StyleChoi, Jiin, and Hee Jin Cho. 2024. "Comprehensive Transcriptomic Profiling of Diverse Brain Tumor Types Uncovers Complex Structures of the Brain Tumor Microenvironment" Biomedicines 12, no. 3: 506. https://doi.org/10.3390/biomedicines12030506
APA StyleChoi, J., & Cho, H. J. (2024). Comprehensive Transcriptomic Profiling of Diverse Brain Tumor Types Uncovers Complex Structures of the Brain Tumor Microenvironment. Biomedicines, 12(3), 506. https://doi.org/10.3390/biomedicines12030506