

Gut Fungal Microbiota Alterations in Pulmonary Arterial Hypertensive Rats

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Groupings
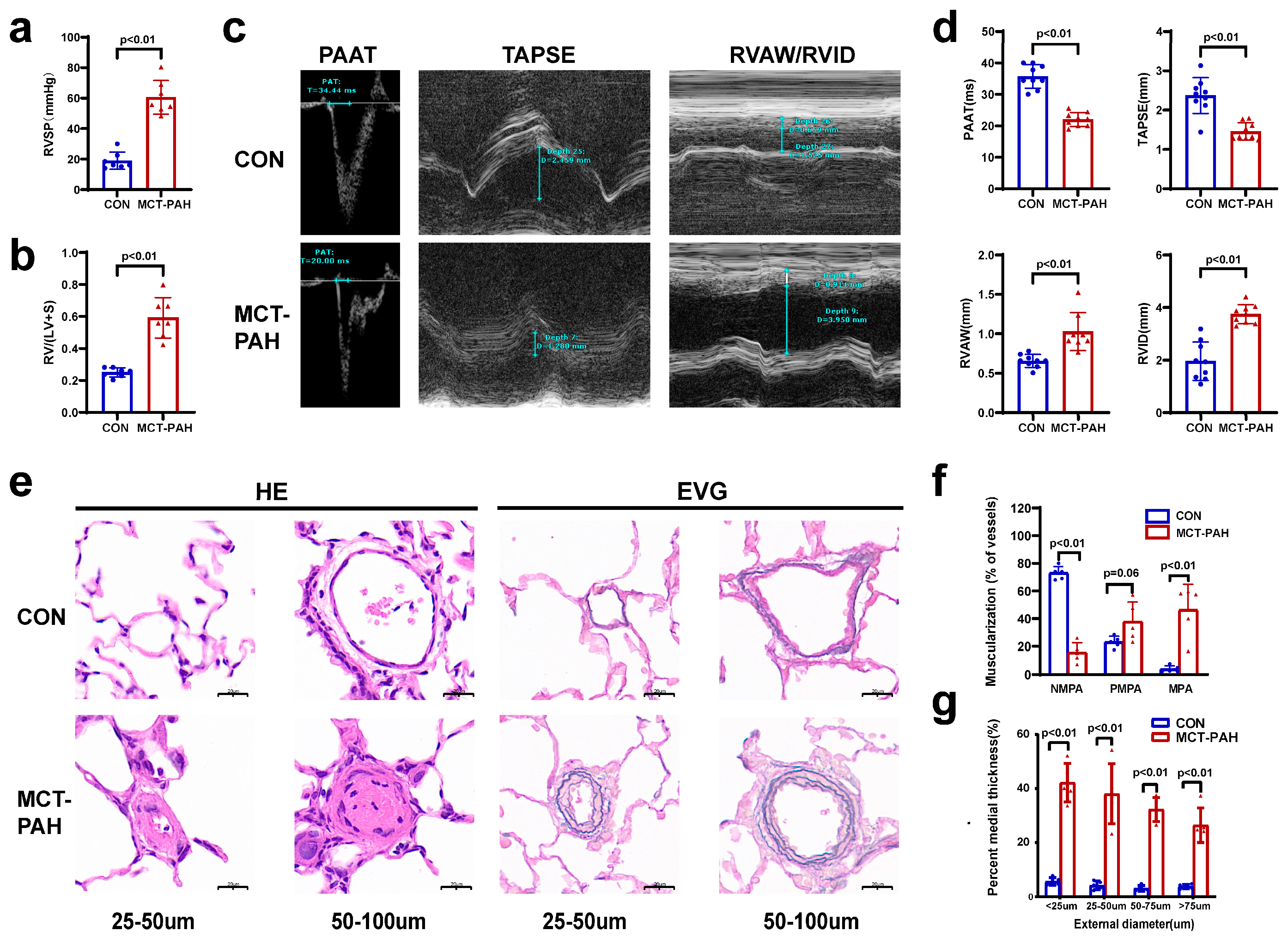
2.2. Echocardiography
2.3. Hemodynamic Measurements and Tissue Processing
2.4. Histological Examination
2.5. Sample Acquisition and DNA Extraction
2.6. Bioinformatics Analysis and Data Analysis
2.7. Statistical Analysis
3. Results
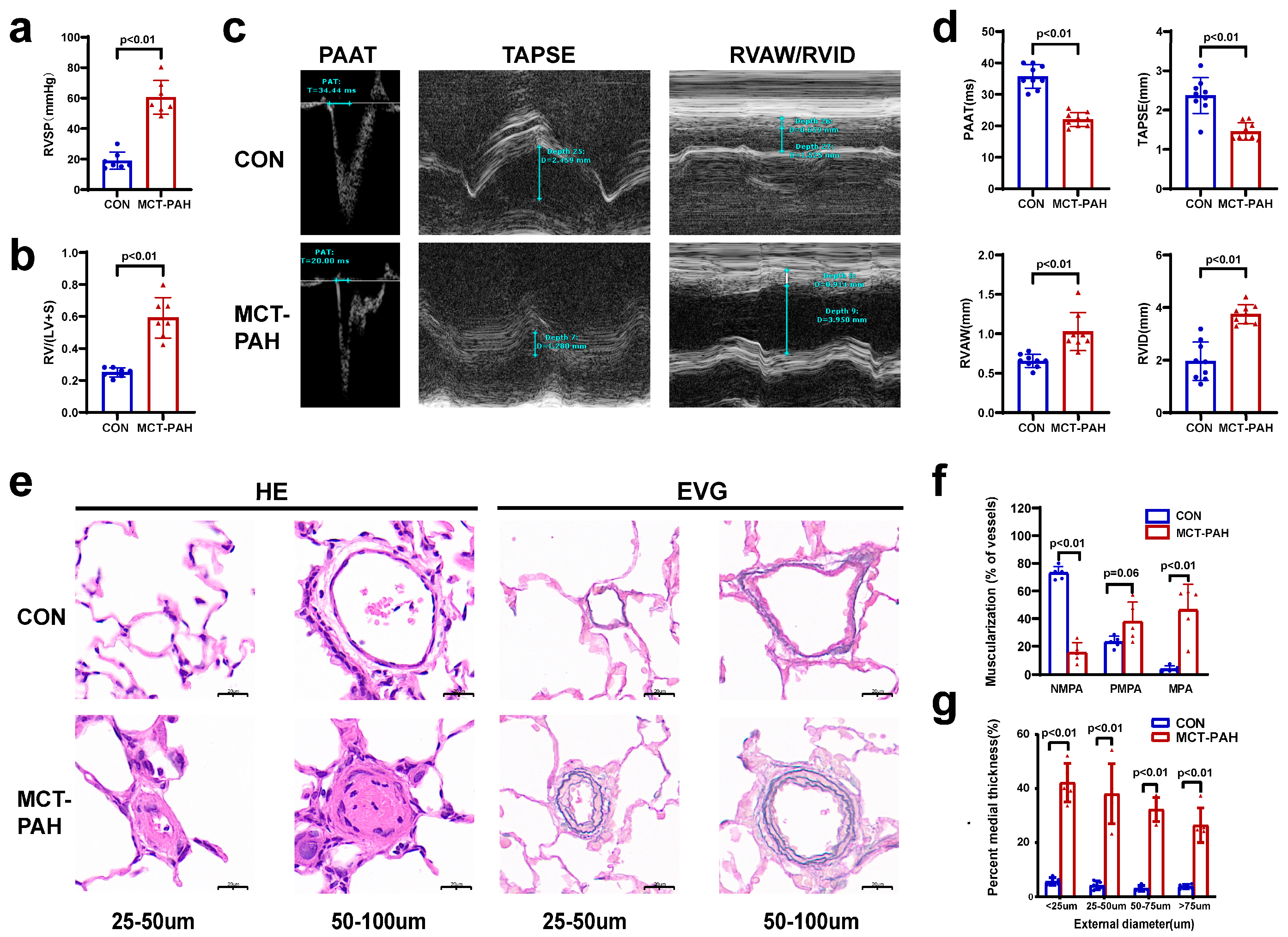
3.1. Severe PAH Developed in Monocrotaline (MCT)-Induced Rats
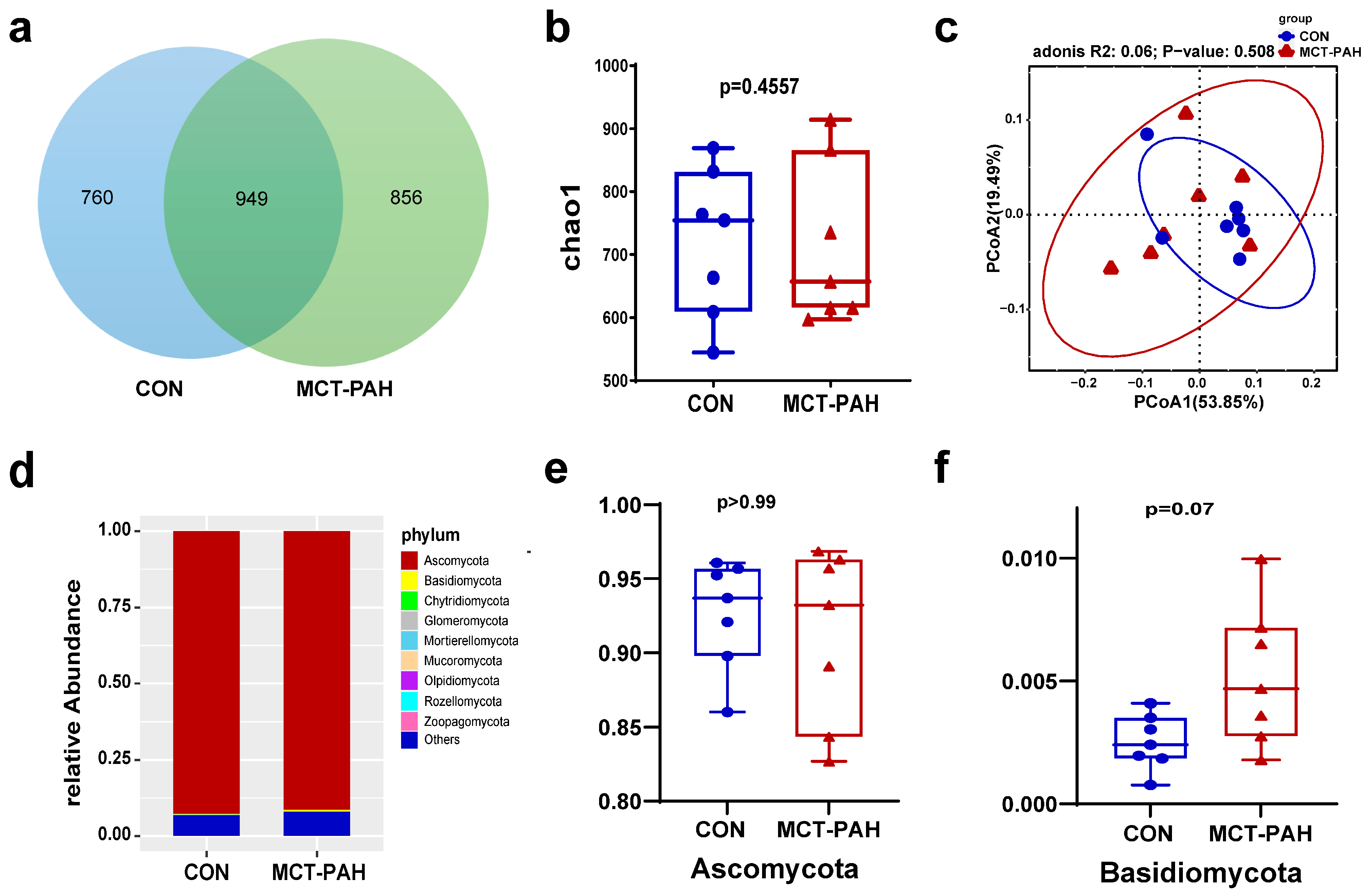
3.2. Characteristics of the Sequence Datasets
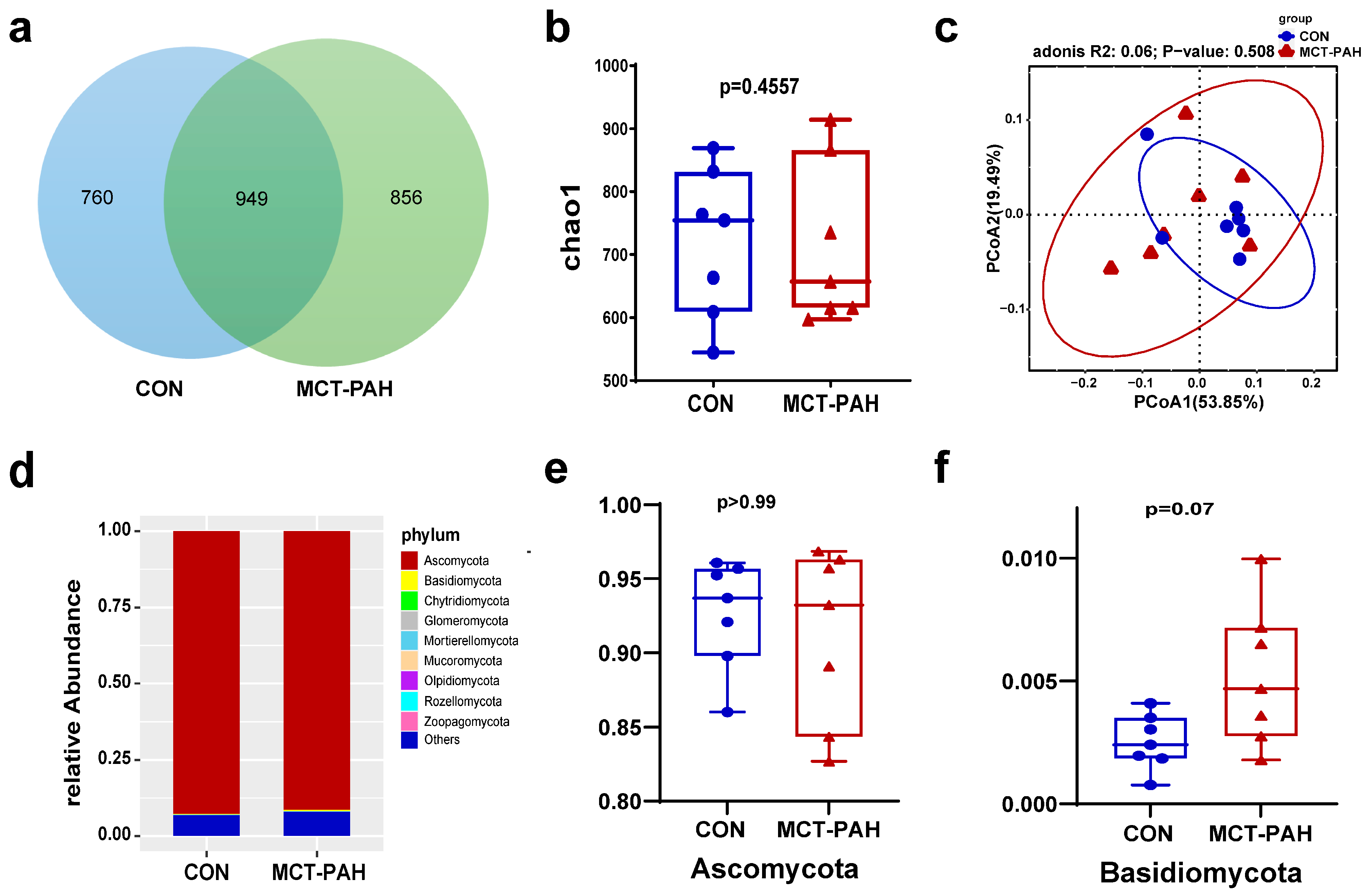
3.3. Ecological Features of the Fecal Fungal Flora in PAH Rats
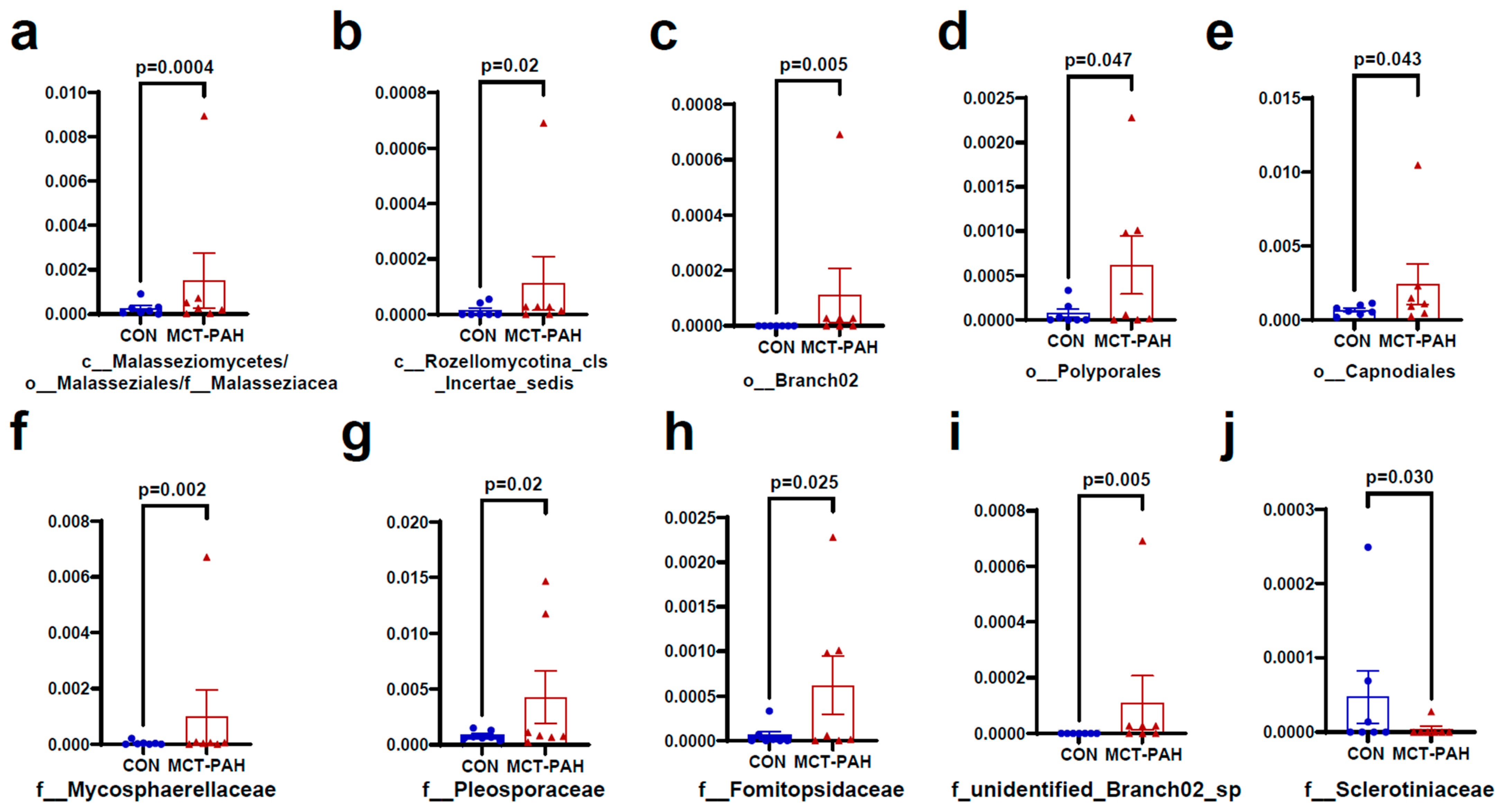
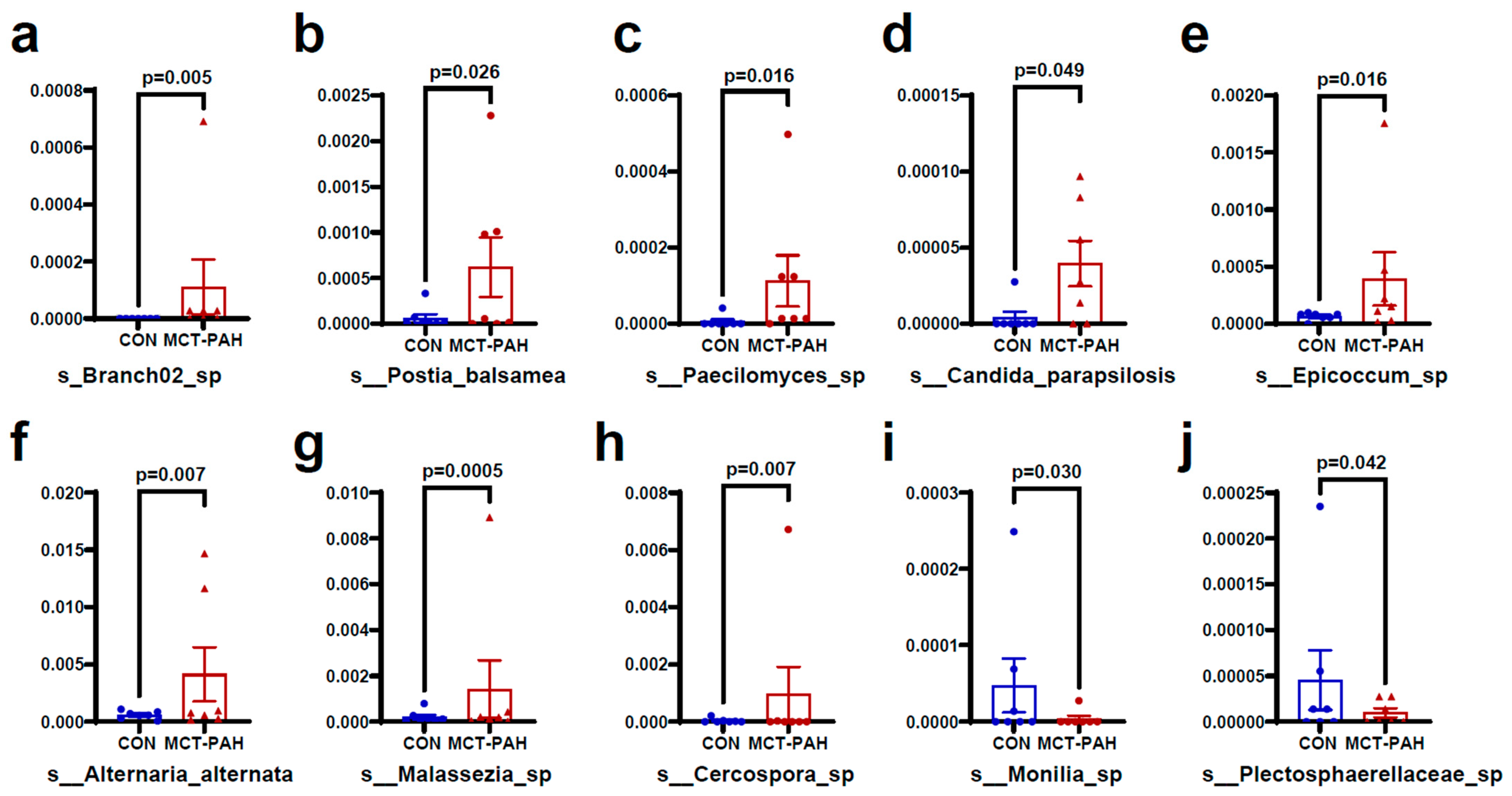
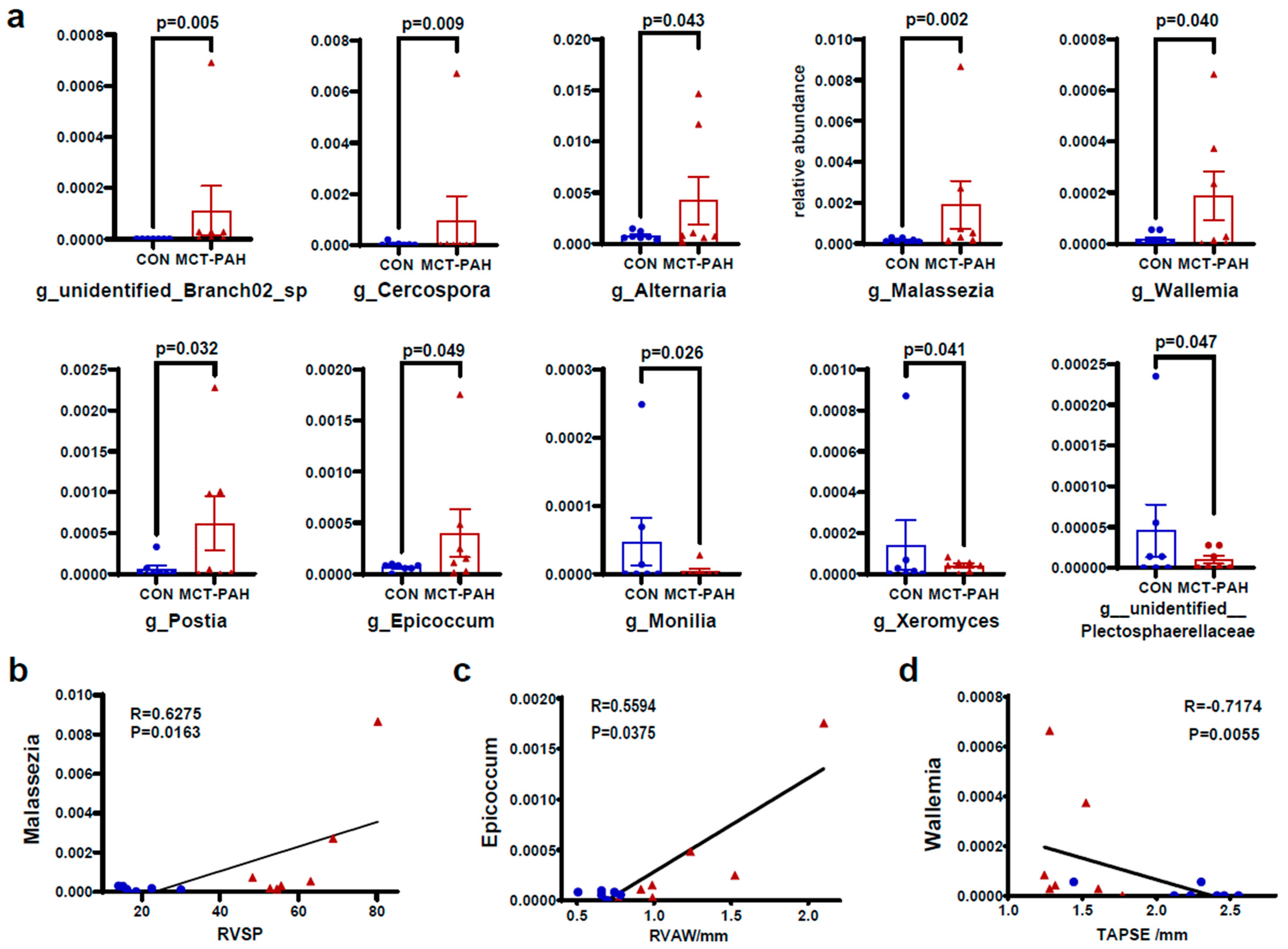
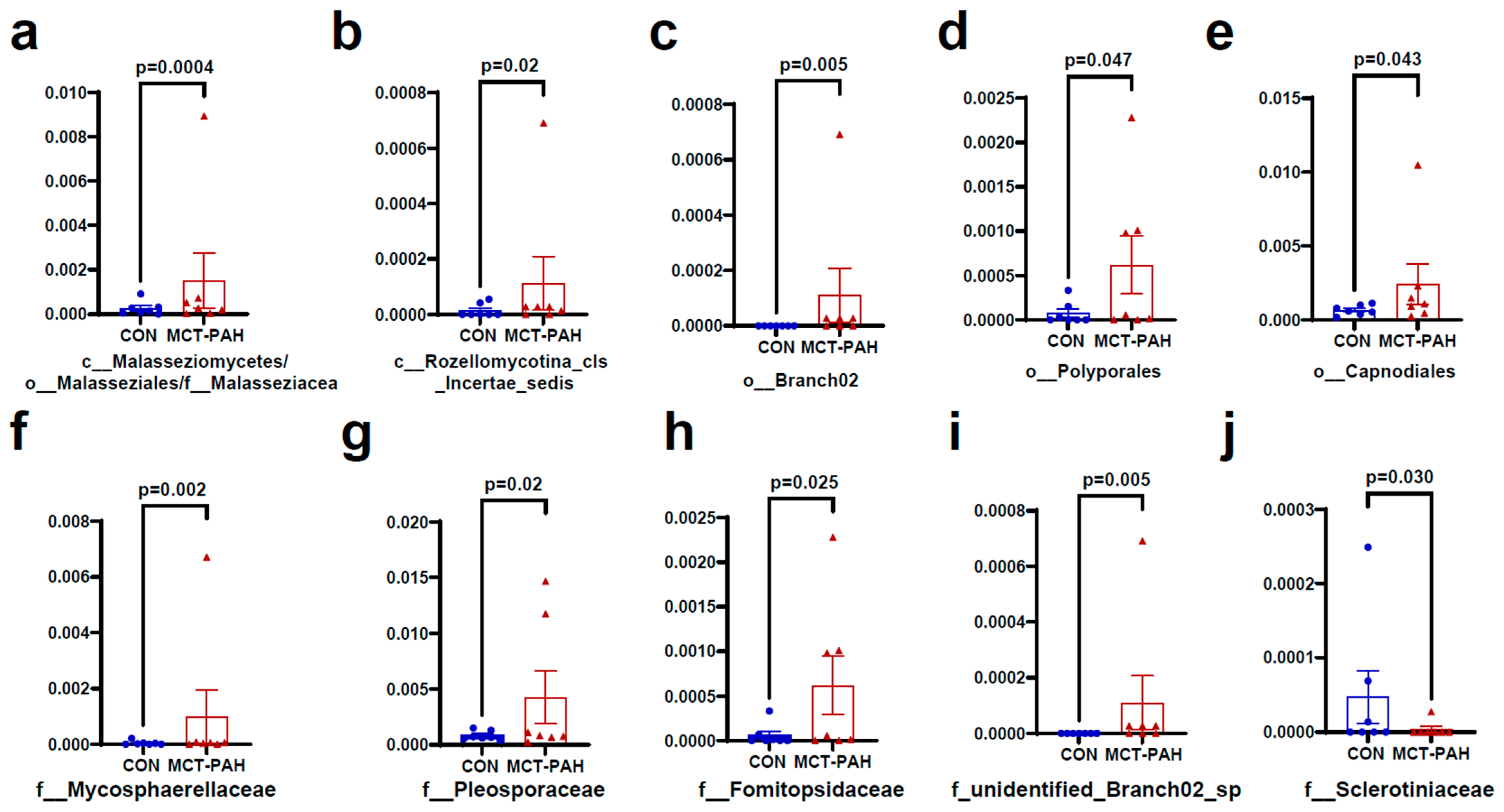
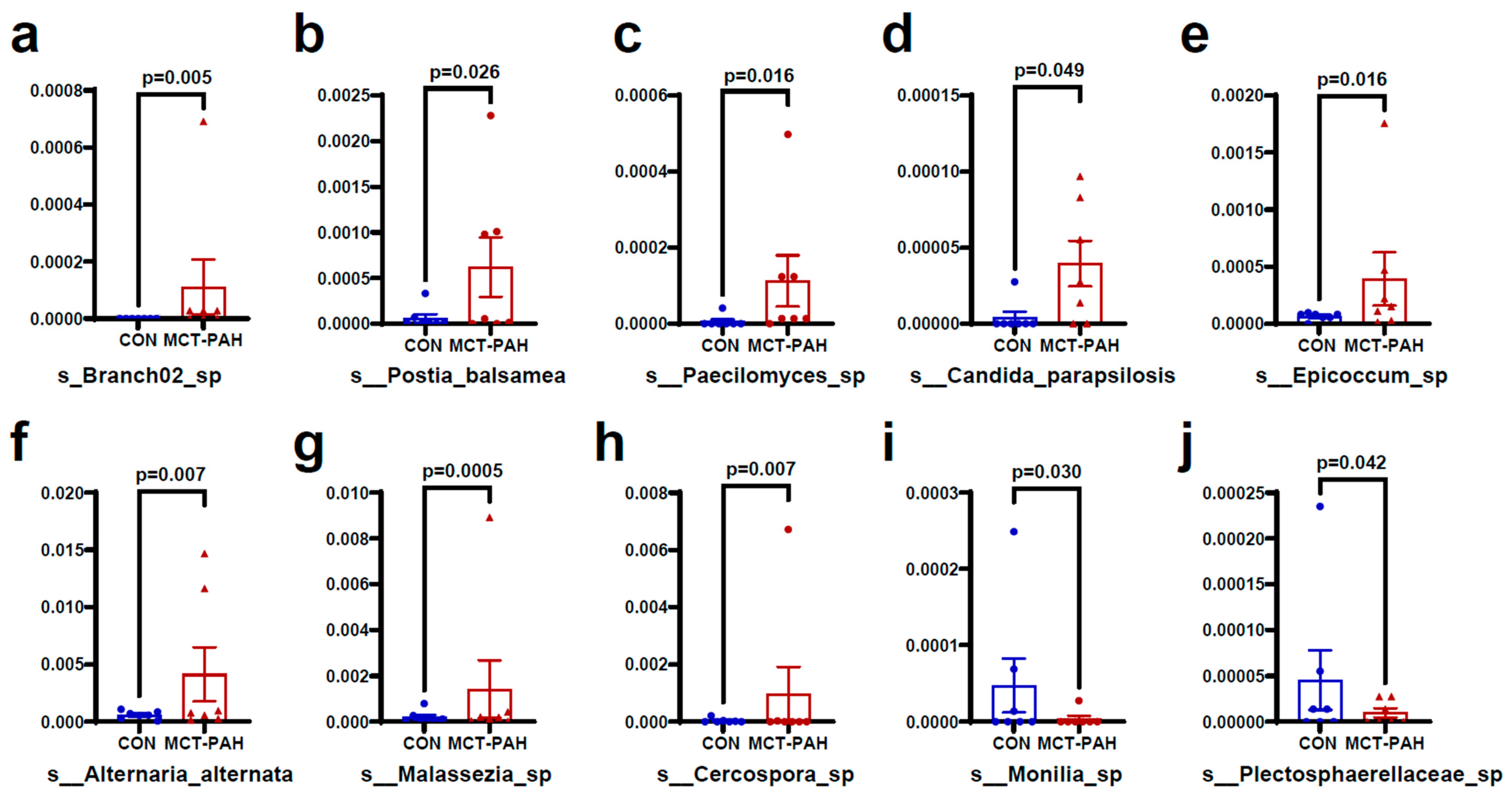
3.4. Alterations of Gut Fungal Taxonomic Compositions in PAH
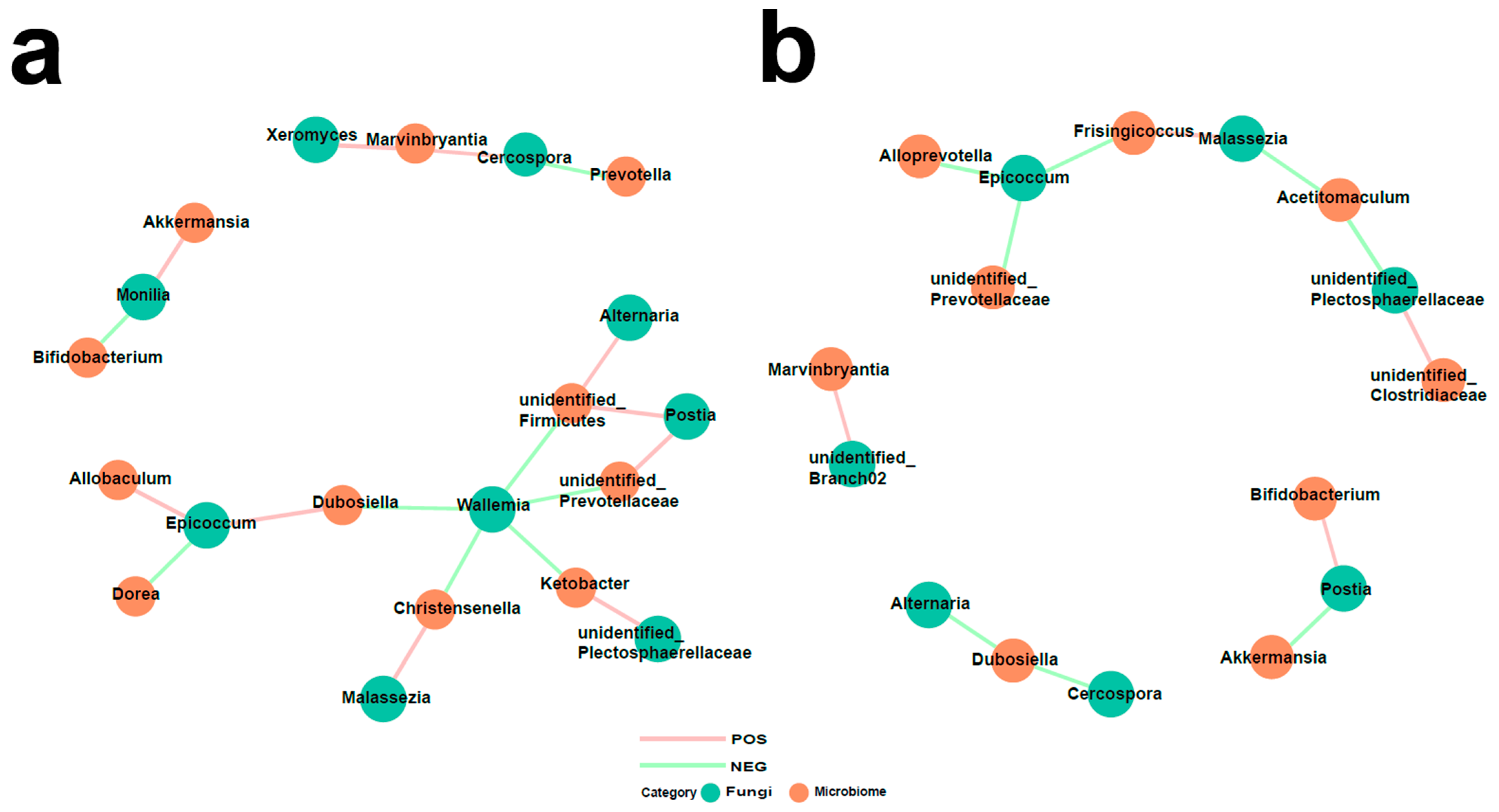
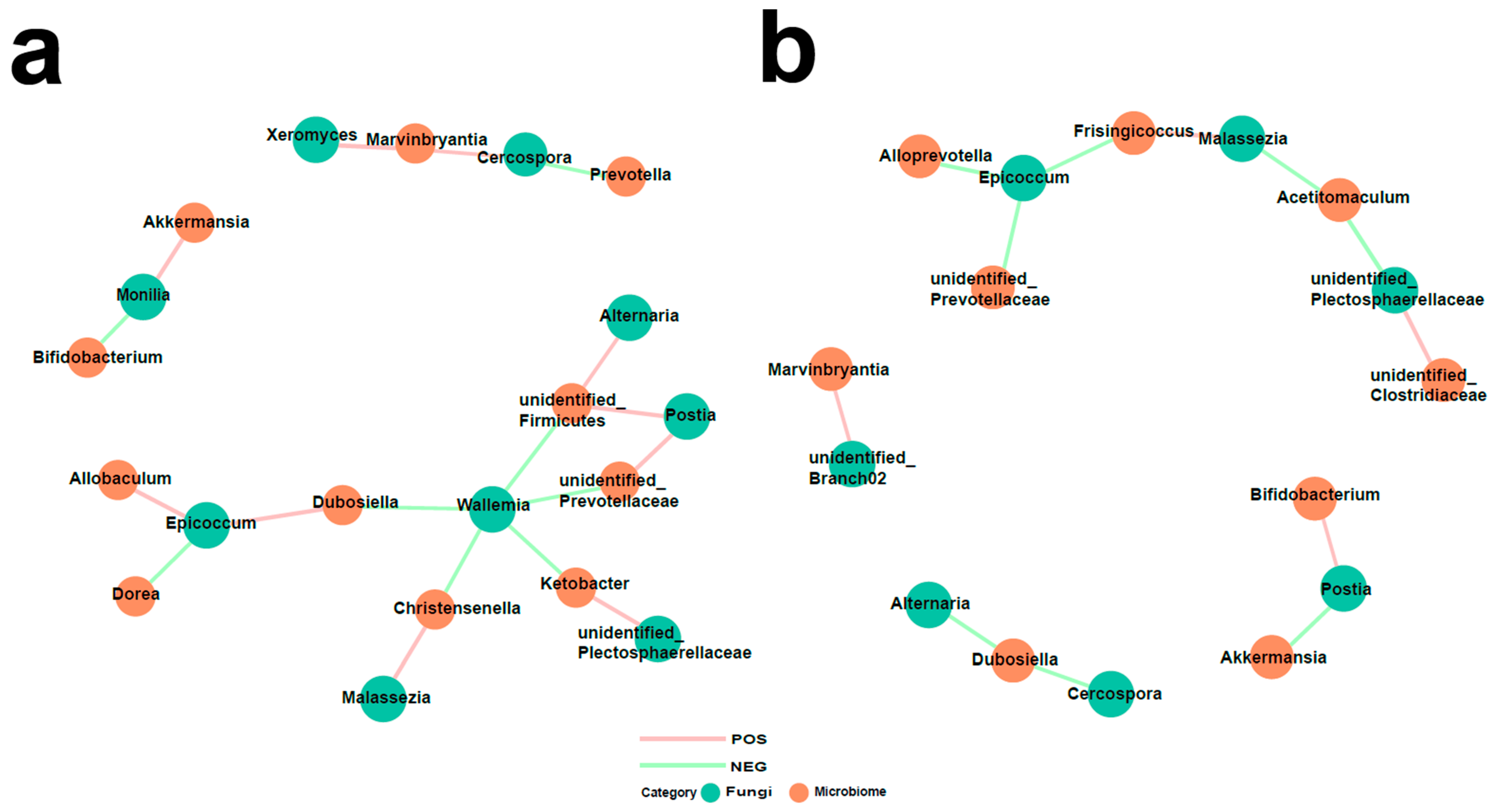
3.5. Interaction of Bacteria and Fungi in Pulmonary Arterial Hypertension
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.; Rigatto, K.; Gazzana, M.B.; Knorst, M.M.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension 2020, 75, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Shui, Y.; Cui, Y.; Tang, C.; Wang, X.; Qiu, X.; Hu, W.; Fei, L.; Li, Y.; Zhang, S.; et al. Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II–induced hypertension. Redox Biol. 2021, 46, 102115. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Strassheim, D.; Sullivan, T.; Verin, A.; Umapathy, N.S.; Dempsey, E.C.; Frank, D.N.; Stenmark, K.R.; Gerasimovskaya, E. The short-chain fatty acid butyrate attenuates pulmonary vascular remodeling and inflammation in hypoxia-induced pul-monary hypertension. Int. J. Mol. Sci. 2021, 22, 9916. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V. Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World J. Gastroenterol. 2010, 16, 2202–2222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Han, B.; Guan, X.; Du, G.; Sheng, B.; Tang, X.; Zhang, Q.; Xie, H.; Jiang, X.; Tan, Q.; et al. Enteric fungi protect against intestinal ischemia–reperfusion injury via inhibiting the SAA1-GSDMD pathway. J. Adv. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, C.; Chai, D.; Li, C.; Qiu, Z.; Kuang, T.; Liu, L.; Deng, W.; Wang, W. Characterization of the intestinal fungal microbiome in patients with hepatocellular carcinoma. J. Transl. Med. 2023, 21, 126. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, A.; Lydia, B.; Huang, C.; Wang, Q.; Yu, Y. Gut mycobiome dysbiosis contributes to the development of hypertension and its response to immunoglobulin light chains. Front. Immunol. 2022, 13, 1089295. [Google Scholar] [CrossRef]
- Wolf, A.J.; Limon, J.J.; Nguyen, C.; Prince, A.; Castro, A.; Underhill, D.M. Malassezia spp. induce inflammatory cytokines and activate NLRP3 inflammasomes in phagocytes. J. Leukoc. Biol. 2021, 109, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Urboniene, D.; Haber, I.; Fang, Y.-H.; Thenappan, T.; Archer, S.L.; Su, J.; Logan, C.C.; Hughes, A.D.; Parker, K.H.; Dhutia, N.M.; et al. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2010, 299, L401–L412. [Google Scholar] [CrossRef] [PubMed]
- Callejo, M.; Mondejar-Parreño, G.; Barreira, B.; Izquierdo-Garcia, J.L.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, Á.; Duarte, J.; Perez-Vizcaino, F. Pulmonary arterial hypertension affects the rat gut microbiome. Sci. Rep. 2018, 8, 9681. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-Y.; Lin, W.-Z.; Li, Y.-L.; Bi, C.; Du, L.-J.; Liu, Y.; Zhou, L.-J.; Liu, T.; Xu, S.; Shi, C.-J.; et al. Roles of oral microbiota and oral-gut microbial transmission in hypertension. J. Adv. Res. 2023, 43, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Mihuta, M.S.; Paul, C.; Borlea, A.; Roi, C.M.; Pescari, D.; Velea-Barta, O.-A.; Mozos, I.; Stoian, D. Connections between serum Trimethylamine N-Oxide (TMAO), a gut-derived metabolite, and vascular biomarkers evaluating arterial stiffness and subclinical atherosclerosis in children with obesity. Front. Endocrinol. 2023, 14, 1253584. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Yuan, W.; Meng, L.-K.; Zhong, J.-C.; Liu, X.-Y. The role and mechanism of gut microbiota in pulmonary arterial hypertension. Nutrients 2022, 14, 4278. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Jayasudha, R.; Das, T.; Kalyana Chakravarthy, S.; Sai Prashanthi, G.; Bhargava, A.; Tyagi, M.; Rani, P.K.; Pappuru, R.R.; Shivaji, S. Gut mycobiomes are altered in people with type 2 diabetes mellitus and diabetic retinopathy. PLoS ONE 2020, 15, e0243077. [Google Scholar] [CrossRef]
- Son, J.S.; Zheng, L.J.; Rowehl, L.M.; Tian, X.; Zhang, Y.; Zhu, W.; Litcher-Kelly, L.; Gadow, K.D.; Gathungu, G.; Robertson, C.E.; et al. Comparison of fecal microbiota in children with autism spectrum disorders and neurotypical siblings in the Simons Simplex Collection. PLoS ONE 2015, 10, e0137725. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, W.; Chen, Y.; Zhao, J.; Wu, S.; Su, X. Flex Meta-Storms elucidates the microbiome local beta-diversity under specific phenotypes. Bioinformatics 2023, 39, btad148. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H.; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Limon, J.J.; Tang, J.; Li, D.; Wolf, A.J.; Michelsen, K.S.; Funari, V.; Gargus, M.; Nguyen, C.; Sharma, P.; Maymi, V.I.; et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe 2019, 25, 377–388.e6. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Li, R.; Fu, R.; Zhang, X.; Yue, B.; Wang, J.; Sun, Z.; Niu, R. Intestinal fungal dysbiosis in mice induced by fluoride. Chemosphere 2020, 245, 125617. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Ali, S.; Shrode, R.L.; Shahi, S.K.; Jensen, S.N.; Hoang, J.; Cassidy, S.; Olalde, H.; Guseva, N.; Paullus, M.; et al. Multiple sclerosis patients have an altered gut mycobiome and increased fungal to bacterial richness. PLoS ONE 2022, 17, e0264556. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hu, M.; Sun, T.; Li, J.; Zhou, Y.; Yan, Y.; Xuan, B.; Wang, J.; Xiong, H.; Ji, L.; et al. Multi-kingdom gut microbiota analyses define bacterial-fungal interplay and microbial markers of pan-cancer immunotherapy across cohorts. Cell Host Microbe 2023, 31, 1930–1943.e4. [Google Scholar] [CrossRef]
- Lee, E.H.; Kim, H.; Koh, J.H.; Cha, K.H.; Lee, K.K.; Kim, W.-U.; Pan, C.-H.; Lee, Y.-H. Dysbiotic but nonpathogenic shift in the fecal mycobiota of patients with rheumatoid arthritis. Gut Microbes 2022, 14, 2149020. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Effective Reads | AvgLen (bp) | GC (%) | Effective (%) |
|---|---|---|---|---|---|---|
| CON1 | 92,726 | 90,560 | 88,665 | 221 | 48.39 | 95.62 |
| CON2 | 82,268 | 81,188 | 79,497 | 222 | 48.39 | 96.63 |
| CON3 | 75,580 | 74,117 | 72,526 | 222 | 48.26 | 95.96 |
| CON4 | 85,737 | 85,089 | 83,417 | 222 | 48.43 | 97.29 |
| CON5 | 85,209 | 83,826 | 82,133 | 226 | 48.16 | 96.39 |
| CON6 | 89,549 | 88,400 | 86,154 | 222 | 48.34 | 96.21 |
| CON7 | 88,015 | 86,300 | 84,055 | 223 | 48.27 | 95.5 |
| MCT1 | 89,709 | 89,113 | 87,205 | 223 | 48.43 | 97.21 |
| MCT2 | 83,547 | 83,056 | 81,245 | 228 | 48.15 | 97.24 |
| MCT3 | 79,204 | 77,489 | 75,775 | 224 | 48.24 | 95.67 |
| MCT4 | 81,053 | 80,264 | 78,504 | 226 | 48.03 | 96.86 |
| MCT5 | 82,838 | 82,001 | 80,003 | 221 | 48.21 | 96.58 |
| MCT6 | 78,948 | 77,663 | 75,731 | 224 | 48.28 | 95.93 |
| MCT7 | 90,372 | 88,565 | 86,191 | 223 | 48.37 | 95.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Meng, L.; Yuan, W.; Gao, Z.; Zhang, X.; Xie, B.; Song, J.; Li, J.; Zhong, J.; Liu, X. Gut Fungal Microbiota Alterations in Pulmonary Arterial Hypertensive Rats. Biomedicines 2024, 12, 298. https://doi.org/10.3390/biomedicines12020298
Chen Y, Meng L, Yuan W, Gao Z, Zhang X, Xie B, Song J, Li J, Zhong J, Liu X. Gut Fungal Microbiota Alterations in Pulmonary Arterial Hypertensive Rats. Biomedicines. 2024; 12(2):298. https://doi.org/10.3390/biomedicines12020298
Chicago/Turabian StyleChen, Yihang, Liukun Meng, Wen Yuan, Zehan Gao, Xun Zhang, Boqia Xie, Jiawei Song, Jifeng Li, Jiuchang Zhong, and Xiaoyan Liu. 2024. "Gut Fungal Microbiota Alterations in Pulmonary Arterial Hypertensive Rats" Biomedicines 12, no. 2: 298. https://doi.org/10.3390/biomedicines12020298
APA StyleChen, Y., Meng, L., Yuan, W., Gao, Z., Zhang, X., Xie, B., Song, J., Li, J., Zhong, J., & Liu, X. (2024). Gut Fungal Microbiota Alterations in Pulmonary Arterial Hypertensive Rats. Biomedicines, 12(2), 298. https://doi.org/10.3390/biomedicines12020298