
Safety Concerns in Neurological Clinical Trials: A Challenge That the FDA Must Resolve


Abstract
1. Introduction
2. Testing Antibodies to Treat Neurological Disorders
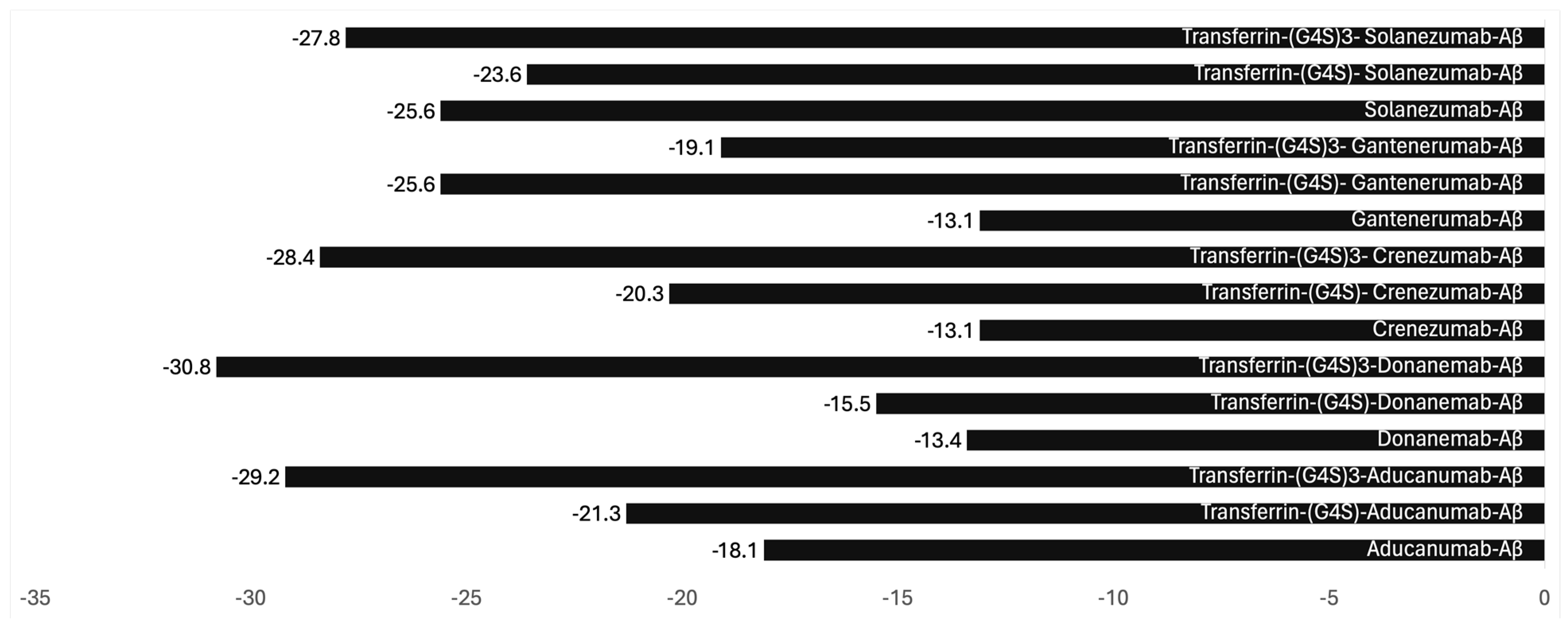
3. The Future
4. Moving Forward
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kessler, W.B.a.C. What Drugmakers Did Not Tell Volunteers in Alzheimer’s Trials. The New York Times. 2024. Available online: https://www.nytimes.com/2024/10/23/health/alzheimers-drug-brain-bleeding.html (accessed on 24 October 2024).
- Giarratana, A.O.; Zheng, C.; Reddi, S.; Teng, S.L.; Berger, D.; Adler, D.; Sullivan, P.; Thakker-Varia, S.; Alder, J. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci. Rep. 2020, 10, 19919. [Google Scholar] [CrossRef]
- Huang, L.K.; Kuan, Y.C.; Lin, H.W.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease: A 2020–2023 update. J. Biomed. Sci. 2023, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 24 October 2024).
- EMA. Leqembi Recommended for Treatment of Early Alzheimer’s Disease, 14 November After Rejection in 2024. Available online: https://www.ema.europa.eu/en/news/leqembi-recommended-treatment-early-alzheimers-disease (accessed on 12 December 2024).
- TGA-Australia. TGA’s Decision to not Register Lecanemab (LEQEMBI). Available online: https://www.tga.gov.au/news/news/tgas-decision-not-register-lecanemab-leqembi (accessed on 24 October 2024).
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Shim, K.H.; Kang, M.J.; Youn, Y.C.; An, S.S.A.; Kim, S. Alpha-synuclein: A pathological factor with Aβ and tau and biomarker in Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 201. [Google Scholar] [CrossRef]
- Mo, J.J.; Li, J.Y.; Yang, Z.; Liu, Z.; Feng, J.S. Efficacy and safety of anti-amyloid-β immunotherapy for Alzheimer’s disease: A systematic review and network meta-analysis. Ann. Clin. Transl. Neurol. 2017, 4, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Baggett, D.; Olson, A.; Parmar, M.S. Novel approaches targeting α-Synuclein for Parkinson’s Disease: Current progress and future directions for the disease-modifying therapies. Brain Disord. 2024, 16, 100163. [Google Scholar] [CrossRef]
- Krämer, J.; Wiendl, H. What Have Failed, Interrupted, and Withdrawn Antibody Therapies in Multiple Sclerosis Taught Us? Neurotherapeutics 2022, 19, 785–807. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Sigurdsson, E.M. Tau Immunotherapies for Alzheimer’s Disease and Related Tauopathies: Progress and Potential Pitfalls. J. Alzheimers Dis. 2018, 64, S555–S565. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Berger, B.; Reichel, E.; Danis, R.P.; Gress, A.; Ye, L.; Magee, M.; Parham, L.R.; McLaughlin, M.M. A Randomized Phase 2 Study of an Anti–Amyloid β Monoclonal Antibody in Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmol. Retin. 2018, 2, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Landen, J.W.; Andreasen, N.; Cronenberger, C.L.; Schwartz, P.F.; Börjesson-Hanson, A.; Östlund, H.; Sattler, C.A.; Binneman, B.; Bednar, M.M. Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 393–401. [Google Scholar] [CrossRef]
- Teng, E.; Manser, P.T.; Pickthorn, K.; Brunstein, F.; Blendstrup, M.; Sanabria Bohorquez, S.; Wildsmith, K.R.; Toth, B.; Dolton, M.; Ramakrishnan, V.; et al. Safety and Efficacy of Semorinemab in Individuals With Prodromal to Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Manoutcharian, K.; Gevorkian, G. Recombinant Antibody Fragments for Immunotherapy of Parkinson’s Disease. BioDrugs 2024, 38, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Bioavailability as Proof to Authorize the Clinical Testing of Neurodegenerative Drugs—Protocols and Advice for the FDA to Meet the ALS Act Vision. Int. J. Mol. Sci. 2024, 25, 10211. [Google Scholar] [CrossRef]
- Kouhi, A.; Pachipulusu, V.; Kapenstein, T.; Hu, P.; Epstein, A.L.; Khawli, L.A. Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives. Int. J. Mol. Sci. 2021, 22, 6442. [Google Scholar] [CrossRef]
- Dumitriu, A.; Popescu, B.O. Placebo effects in neurological diseases. J. Med. Life 2010, 3, 114–121. [Google Scholar] [PubMed]
- Oken, B.S. Placebo effects: Clinical aspects and neurobiology. Brain 2008, 131, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Peciña, M.; Zubieta, J.-K. Molecular Mechanisms of Placebo Responses In Humans. Mol. Psychiatry 2014, 20, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. A Modern View of Placebo Interventions: From Comparative Clinical Trials to Novel Therapies to Quantum Tunnelling. Preprints 2024. [CrossRef]
- Niazi, S.K.; Mariam, Z.; Magoola, M. Engineered Antibodies to Improve Efficacy against Neurodegenerative Disorders. Int. J. Mol. Sci. 2024, 25, 6683. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Non-Invasive Drug Delivery across the Blood–Brain Barrier: A Prospective Analysis. Pharmaceutics 2023, 15, 2599. [Google Scholar] [CrossRef] [PubMed]
- Tien, J.; Leonoudakis, D.; Petrova, R.; Trinh, V.; Taura, T.; Sengupta, D.; Jo, L.; Sho, A.; Yun, Y.; Doan, E.; et al. Modifying antibody-FcRn interactions to increase the transport of antibodies through the blood-brain barrier. MAbs 2023, 15, 2229098. [Google Scholar] [CrossRef] [PubMed]
- Marathe, P.H.; Shyu, W.C.; Humphreys, W.G. The use of radiolabeled compounds for ADME studies in discovery and exploratory development. Curr. Pharm. Des. 2004, 10, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [PubMed]
- Nelke, C.; Spatola, M.; Schroeter, C.B.; Wiendl, H.; Lünemann, J.D. Neonatal Fc Receptor-Targeted Therapies in Neurology. Neurotherapeutics 2022, 19, 729–740. [Google Scholar] [CrossRef]
- Watanabe, T.; Arashida, N.; Fujii, T.; Shikida, N.; Ito, K.; Shimbo, K.; Seki, T.; Iwai, Y.; Hirama, R.; Hatada, N.; et al. Exo-Cleavable Linkers: Enhanced Stability and Therapeutic Efficacy in Antibody–Drug Conjugates. J. Med. Chem. 2024, 67, 18124–18138. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Li, Y.; Feola, M.; Guerra, A.; Casu, C.; Prasad, P.; Mammen, L.; Ali, F.; Vaicikauskas, E.; Rivella, S.; et al. Lobe specificity of iron binding to transferrin modulates murine erythropoiesis and iron homeostasis. Blood 2019, 134, 1373–1384. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Neely, M.D.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M.; et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 054124. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, Á.; Tanaka, K.; Niwa, M. A new blood–brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Hynynen, K. Noninvasive and targeted drug delivery to the brain using focused ultrasound. ACS Chem. Neurosci. 2013, 4, 519–526. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef]
- Nemes, P.; Vertes, A. Laser Ablation Electrospray Ionization for Atmospheric Pressure, in Vivo, and Imaging Mass Spectrometry. Anal. Chem. 2007, 79, 8098–8106. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Nie, W.; Zhang, J.; Xie, H.Y. Cell-Membrane-Based Biomimetic Systems with Bioorthogonal Functionalities. Acc. Chem. Res. 2020, 53, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xie, J.; Yan, R.; Yu, Z.; Sun, Z.; Yu, F.; Gong, X.; Feng, H.; Lu, J.; Zhang, Y. A pilot study of pancreatic islet amyloid PET imaging with [18F]FDDNP. Nucl. Med. Commun. 2018, 39, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.G.; Andrieș, G.; Căinap, C.; Chiș, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Egleton, R.D. Pathophysiology of the blood-brain barrier: Animal models and methods. Curr. Top. Dev. Biol. 2008, 80, 277–309. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Chen, C.; Lu, Z.L.; Dong, Q. Brain Imaging Techniques and Their Applications in Decision-Making Research. Xin Li Xue Bao 2010, 42, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Ross, B.; Lin, A. Magnetic resonance spectroscopy in neurological diagnosis. Neurol. Clin. 2009, 27, 21–60, xiii. [Google Scholar] [CrossRef]
- Appelt-Menzel, A.; Oerter, S.; Mathew, S.; Haferkamp, U.; Hartmann, C.; Jung, M.; Neuhaus, W.; Pless, O. Human iPSC-Derived Blood-Brain Barrier Models: Valuable Tools for Preclinical Drug Discovery and Development? Curr. Protoc. Stem Cell Biol. 2020, 55, e122. [Google Scholar] [CrossRef]
- Proetzel, G.; Wiles, M.V.; Roopenian, D.C. Genetically engineered humanized mouse models for preclinical antibody studies. BioDrugs 2014, 28, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Wong, W. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 2020, 26, S177–S183. [Google Scholar] [CrossRef] [PubMed]
- Regulations, U.C.o.F. PART 46—Protection of Human Subjects. Authority:5 U.S.C. 301; 42 U.S.C. 289(a); 42 U.S.C. 300v-1(b). Available online: https://www.ecfr.gov/current/title-45/subtitle-A/subchapter-A/part-461982 (accessed on 24 October 2024).
- Regulations, U.C.o.F. 312.42 Clinical Holds and Requests for Modification. Available online: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312/subpart-C/section-312.422004 (accessed on 24 October 2024).
- World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Monoclonal Antibody | Target | Indication | Stages |
|---|---|---|---|
| Aducanumab [10] | Amyloid-β | Alzheimer’s Disease | Initially approved, then withdrawn due to efficacy concerns and safety issues. |
| Bapineuzumab [15] | Amyloid-β | Alzheimer’s Disease | Phase III trials did not demonstrate significant cognitive improvements; development was discontinued. |
| Bepranemab [16] | Tau | Alzheimer’s Disease | Phase II trials did not meet primary endpoints; the development status was uncertain. |
| Crenezumab [17] | Amyloid-β | Alzheimer’s Disease | Phase III trials did not meet primary endpoints; development was discontinued. |
| Gantenerumab [18], | Amyloid-β | Alzheimer’s Disease | Phase III trials did not meet primary endpoints; development was discontinued. |
| GSK933776 [19] | Amyloid-β | Alzheimer’s Disease | Phase II trials did not meet primary endpoints; development was discontinued. |
| Ponezumab [20] | Amyloid-β | Alzheimer’s Disease | Phase II trials showed no significant efficacy; development halted. |
| Semorinema [21] | Tau | Alzheimer’s Disease | Phase II trials did not meet primary endpoints; the development status was uncertain. |
| Solanezumab [22] | Amyloid-β | Alzheimer’s Disease | Phase III trials failed to meet primary endpoints; development halted. |
| Tilavonemab [16] | Tau | Alzheimer’s Disease | Phase II trials did not meet primary endpoints; development status is uncertain. |
| Cinpanemab [23], | α-Synuclein | Parkinson’s Disease | Phase II SPARK trial terminated due to lack of efficacy. |
| Prasinezumab | α-Synuclein | Parkinson’s Disease | Phase II trials did not meet primary endpoints; the development status was uncertain. |
| Antibody | United States | European Union | United Kingdom | Japan | China | Australia | Canada |
|---|---|---|---|---|---|---|---|
| Aducanumab | Approved by the FDA (2021); FDA | Rejected by the EMA (2021); EMA | Not approved | Not approved | Not approved | Not approved | Not approved |
| Lecanemab | Approved by the FDA (2023); FDA | Rejected by the EMA (2023); Reuters | Approved by the MHRA (2023); not recommended by NICE; Reuters | Approved (2023) | Approved (2023) | Not approved by TGA (2023); Herald Sun | Approved (2023) |
| Donanemab | Approved by the FDA (2024); FDA | Under EMA review | Approved by the MHRA (2024); not recommended by NICE for NHS; Reuters | Application submitted, pending decision | Application submitted, pending decision | Application submitted, pending decision | Application submitted, pending decision |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K. Safety Concerns in Neurological Clinical Trials: A Challenge That the FDA Must Resolve. Biomedicines 2024, 12, 2918. https://doi.org/10.3390/biomedicines12122918
Niazi SK. Safety Concerns in Neurological Clinical Trials: A Challenge That the FDA Must Resolve. Biomedicines. 2024; 12(12):2918. https://doi.org/10.3390/biomedicines12122918
Chicago/Turabian StyleNiazi, Sarfaraz K. 2024. "Safety Concerns in Neurological Clinical Trials: A Challenge That the FDA Must Resolve" Biomedicines 12, no. 12: 2918. https://doi.org/10.3390/biomedicines12122918
APA StyleNiazi, S. K. (2024). Safety Concerns in Neurological Clinical Trials: A Challenge That the FDA Must Resolve. Biomedicines, 12(12), 2918. https://doi.org/10.3390/biomedicines12122918