

Evaluating the Efficacy of Levetiracetam on Non-Cognitive Symptoms and Pathology in a Tau Mouse Model


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.2.1. Short Term Study
2.2.2. Long-Term Study
2.3. Behavioral Testing
2.4. Tissue Collection
2.5. Histopathology
2.6. Biochemical Analyses
2.7. Quantification of Levetiracetam in Biological Samples
2.8. Statistical Analysis and Rigor
3. Results
3.1. Reduction in Non-Cognitive Symptoms by Pharmacotropic Drugs in a Mouse Model of Tauopathy
3.2. Decreased Body Weight and Food Intake in Non-Transgenic but Not Tg4510 Mice, Following 3 Months of Diet with Levetiracetam
3.3. Behavior Analysis
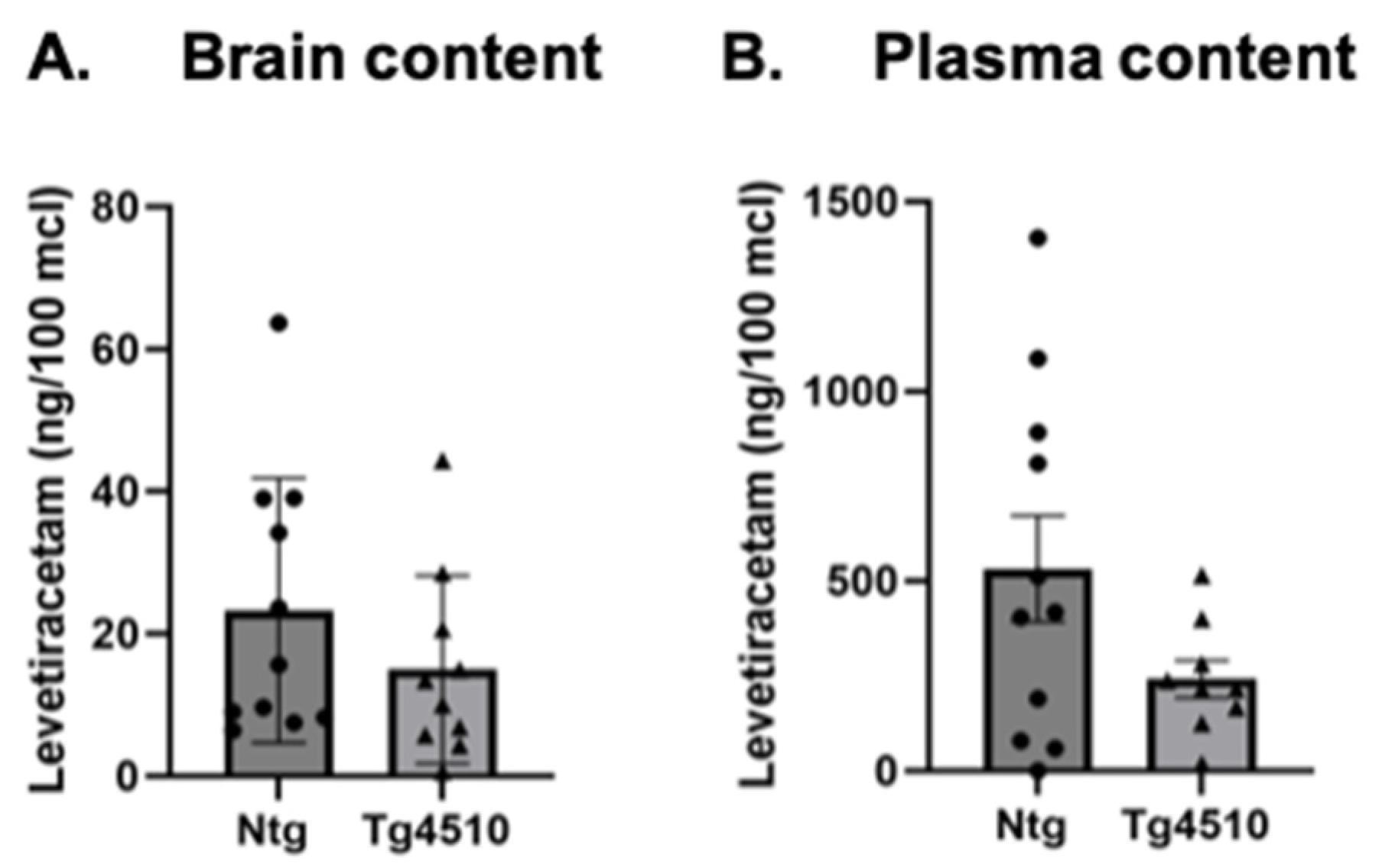
3.4. Levetiracetam Content
3.5. Treatment with Levetiracetam Induced a Mild Decrease in Phosphorylated Tau
3.6. Levetiracetam Normalized Levels of SV2A in Tau Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef]
- Wainaina, M.N.; Chen, Z.; Zhong, C. Environmental factors in the development and progression of late-onset Alzheimer’s disease. Neurosci. Bull. 2014, 30, 253–270. [Google Scholar] [CrossRef]
- Surguchov, A.; Emamzadeh, F.N.; Titova, M.; Surguchev, A.A. Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era. Biomedicines 2023, 11, 1215. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Baudic, S.; Barba, G.D.; Thibaudet, M.C.; Smagghe, A.; Remy, P.; Traykov, L. Executive function deficits in early Alzheimer’s disease and their relations with episodic memory. Arch. Clin. Neuropsychol. 2006, 21, 15–21. [Google Scholar] [CrossRef] [PubMed]
- White, H.; Pieper, C.; Schmader, K. The association of weight change in Alzheimer’s disease with severity of disease and mortality: A longitudinal analysis. J. Am. Geriatr. Soc. 1998, 46, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- White, H.K.; McConnell, E.S.; Bales, C.W.; Kuchibhatla, M. A 6-month observational study of the relationship between weight loss and behavioral symptoms in institutionalized Alzheimer’s disease subjects. J. Am. Med. Dir. Assoc. 2004, 5, 89–97. [Google Scholar] [CrossRef]
- Gillette, G.S.; Abellan, V.K.; Alix, E.; Andrieu, S.; Belmin, J.; Berrut, G.; Bonnefoy, M.; Brocker, P.; Constans, T.; Ferry, M.; et al. IANA (International Academy on Nutrition and Aging) Expert Group: Weight loss and Alzheimer’s disease. J. Nutr. Health Aging. 2007, 11, 38–48. [Google Scholar]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Gillette-Guyonnet, S.; Nourhashemi, F.; Andrieu, S.; de Glisezinski, I.; Ousset, P.J.; Riviere, D.; Albarede, J.L.; Vellas, B. Weight loss in Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 637s–642s. [Google Scholar] [CrossRef] [PubMed]
- Guerin, O.; Soto, M.E.; Brocker, P.; Robert, P.H.; Benoit, M.; Vellas, B. Nutritional status assessment during Alzheimer’s disease: Results after one year (the REAL French Study Group). J. Nutr. Health Aging 2005, 9, 81–84. [Google Scholar]
- Gillette-Guyonnet, S.; Cortes, F.; Cantet, C.; Vellas, B. Long-term cholinergic treatment is not associated with greater risk of weight loss during Alzheimer’s disease: Data from the French REAL.FR cohort. J. Nutr. Health Aging 2005, 9, 69–73. [Google Scholar] [PubMed]
- Guerin, O.; Andrieu, S.; Schneider, S.M.; Milano, M.; Boulahssass, R.; Brocker, P.; Vellas, B. Different modes of weight loss in Alzheimer disease: A prospective study of 395 patients. Am. J. Clin. Nutr. 2005, 82, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Lauque, S.; Gillette-Guyonnet, S.; Andrieu, S.; Cortes, F.; Nourhashemi, F.; Cantet, C.; Ousset, P.J.; Grandjean, H. Impact of nutritional status on the evolution of Alzheimer’s disease and on response to acetylcholinesterase inhibitor treatment. J. Nutr. Health Aging 2005, 9, 75–80. [Google Scholar] [PubMed]
- Faxen-Irving, G.; Basun, H.; Cederholm, T. Nutritional and cognitive relationships and long-term mortality in patients with various dementia disorders. Age Ageing 2005, 34, 136–141. [Google Scholar] [CrossRef]
- Andrieu, S.; Reynish, W.; Nourhashemi, F.; Ousset, P.J.; Grandjean, H.; Grand, A.; Albarede, J.L.; Vellas, B. Nutritional risk factors for institutional placement in Alzheimer’s disease after one year follow-up. J. Nutr. Health Aging 2001, 5, 113–117. [Google Scholar]
- Gambassi, G.; Landi, F.; Lapane, K.L.; Sgadari, A.; Mor, V.; Bernabei, R. Predictors of mortality in patients with Alzheimer’s disease living in nursing homes. J. Neurol. Neurosurg. Psychiatry 1999, 67, 59–65. [Google Scholar] [CrossRef]
- Devanand, D.P. Behavioral complications and their treatment in Alzheimer’s disease. Geriatrics 1997, 52 (Suppl. S2), S37–S39. [Google Scholar]
- Pugh, P.L.; Richardson, J.C.; Bate, S.T.; Upton, N.; Sunter, D. Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer’s disease. Behav. Brain Res. 2007, 178, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Vloeberghs, E.; Van Dam, D.; Franck, F.; Serroyen, J.; Geert, M.; Staufenbiel, M.; De Deyn, P.P. Altered ingestive behavior, weight changes, and intact olfactory sense in an APP overexpression model. Behav. Neurosci. 2008, 122, 491–497. [Google Scholar] [CrossRef]
- Morgan, D.; Gordon, M.N. Amyloid, hyperactivity, and metabolism: Theoretical comment on Vloeberghs et al. (2008). Behav. Neurosci. 2008, 122, 730–732. [Google Scholar] [CrossRef]
- Ishii, M.; Wang, G.; Racchumi, G.; Dyke, J.P. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 2014, 34, 9096–9106. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef]
- SantaCruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; Marín de Evsikova, C.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Hauser, W.A.; Annegers, J.F.; Kokmen, E.; Rocca, W.A. Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727–730. [Google Scholar] [CrossRef]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef]
- Costa, C.; Nardi Cesarini, E.; Eusebi, P.; Franchini, D.; Casucci, P.; De Giorgi, M.F.; Calvello, C.; Paolini Paoletti, F.; Romoli, M.; Parnetti, L. Incidence and Risk Factors Epilepsy in Patients with Dementia: A Population-Based Study Using Regional Healthcare Databases in Umbria. J. Alzheimer’s Dis. 2024, 98, 1533–1542. [Google Scholar] [CrossRef]
- Del Pozo, A.; Knox, K.M.; Lehmann, L.M.; Davidson, S.; Rho, S.L.; Jayadev, S.; Barker-Haliski, M. Chronic evoked seizures in young pre-symptomatic APP/PS1 mice induce serotonin changes and accelerate onset of Alzheimer’s disease-related neuropathology. Prog. Neurobiol. 2024, 235, 102591. [Google Scholar] [CrossRef] [PubMed]
- Dejakaisaya, H.; Kwan, P.; Jones, N.C. Astrocyte and glutamate involvement in the pathogenesis of epilepsy in Alzheimer’s disease. Epilepsia 2021, 62, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Saccaro, L.F.; Busceti, C.L.; Biagioni, F.; Fornai, F. Epilepsy and Alzheimer’s Disease: Potential mechanisms for an association. Brain Res. Bull. 2020, 160, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Besli, G.E.; Yuksel Karatoprak, E.; Yilmaz, S. Efficacy and safety profile of intravenous levetiracetam versus phenytoin in convulsive status epilepticus and acute repetitive seizures in children. Epilepsy Behav. 2020, 111, 107289. [Google Scholar] [CrossRef] [PubMed]
- Glauser, T.A.; Pellock, J.M.; Bebin, E.M.; Fountain, N.B.; Ritter, F.J.; Jensen, C.M.; Shields, W.D. Efficacy and safety of levetiracetam in children with partial seizures: An open-label trial. Epilepsia 2002, 43, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Ben-Menachem, E.; Falter, U. Efficacy and tolerability of levetiracetam 3000 mg/d in patients with refractory partial seizures: A multicenter, double-blind, responder-selected study evaluating monotherapy. European Levetiracetam Study Group. Epilepsia 2000, 41, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Choi, H.; Hirsch, L.J.; Katz, A.; Legge, A.; Buchsbaum, R.; Detyniecki, K. Psychiatric and behavioral side effects of antiepileptic drugs in adults with epilepsy. Epilepsy Behav. EB 2017, 76, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, L.; Tang, X. Levetiracetam is associated with decrease in subclinical epileptiform discharges and improved cognitive functions in pediatric patients with autism spectrum disorder. Neuropsychiatr. Dis. Treat. 2017, 13, 2321–2326. [Google Scholar] [CrossRef]
- Solaro, C.; de Sire, A.; Messmer Uccelli, M.; Mueller, M.; Bergamaschi, R.; Gasperini, C.; Restivo, D.A.; Stabile, M.R.; Patti, F. Efficacy of levetiracetam on upper limb movement in multiple sclerosis patients with cerebellar signs: A multicenter double-blind, placebo-controlled, crossover study. Eur. J. Neurol. 2020, 27, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Awaad, Y.; Michon, A.M.; Minarik, S. Use of levetiracetam to treat tics in children and adolescents with Tourette syndrome. Mov. Disord. Off. J. Mov. Disord. Soc. 2005, 20, 714–718. [Google Scholar] [CrossRef]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef]
- Shi, J.; Anderson, D.; Lynch, B.A.; Castaigne, J.-G.; Foerch, P.; Lebon, F. Combining modelling and mutagenesis studies of synaptic vesicle protein 2A to identify a series of residues involved in racetam binding. Biochem. Soc. Trans. 2011, 39, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Crowder, K.M.; Gunther, J.M.; Jones, T.A.; Hale, B.D.; Zhang, H.Z.; Peterson, M.R.; Scheller, R.H.; Chavkin, C.; Bajjalieh, S.M. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. USA 1999, 96, 15268–15273. [Google Scholar] [CrossRef] [PubMed]
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B Function as Redundant Ca2+ Regulators in Neurotransmitter Release. Neuron 1999, 24, 1003–1016. [Google Scholar] [CrossRef]
- Niespodziany, I.; Klitgaard, H.; Margineanu, D.G. Levetiracetam inhibits the high-voltage-activated Ca2+ current in pyramidal neurones of rat hippocampal slices. Neurosci. Lett. 2001, 306, 5–8. [Google Scholar] [CrossRef]
- Yan, H.-D.; Ishihara, K.; Seki, T.; Hanaya, R.; Kurisu, K.; Arita, K.; Serikawa, T.; Sasa, M. Inhibitory effects of levetiracetam on the high-voltage-activated L-type Ca2+ channels in hippocampal CA3 neurons of spontaneously epileptic rat (SER). Brain Res. Bull. 2013, 90, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Nowack, A.; Malarkey, E.B.; Yao, J.; Bleckert, A.; Hill, J.; Bajjalieh, S.M. Levetiracetam Reverses Synaptic Deficits Produced by Overexpression of SV2A. PLoS ONE 2011, 6, e29560. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.-M.; Hannestad, J.; Holden, D.; Kervyn, S.; Nabulsi, N.; Tytgat, D.; Huang, Y.; Chanteux, H.; Staelens, L.; Matagne, A.; et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia 2016, 57, 201–209. [Google Scholar] [CrossRef]
- Meehan, A.L.; Yang, X.; McAdams, B.D.; Yuan, L.; Rothman, S.M. A new mechanism for antiepileptic drug action: Vesicular entry may mediate the effects of levetiracetam. J. Neurophysiol. 2011, 106, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Daniels, V.; Wood, M.; Leclercq, K.; Kaminski, R.M.; Gillard, M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br. J. Pharmacol. 2013, 169, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Stockburger, C.; Miano, D.; Baeumlisberger, M.; Pallas, T.; Arrey, T.N.; Karas, M.; Friedland, K.; Müller, W.E. A Mitochondrial Role of SV2a Protein in Aging and Alzheimer’s Disease: Studies with Levetiracetam. J. Alzheimer’s Dis. 2016, 50, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Rigo, J.-M.; Hans, G.; Nguyen, L.; Rocher, V.; Belachew, S.; Malgrange, B.; Leprince, P.; Moonen, G.; Selak, I.; Matagne, A.; et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. Br. J. Pharmacol. 2002, 136, 659–672. [Google Scholar] [CrossRef]
- Carunchio, I.; Pieri, M.; Ciotti, M.T.; Albo, F.; Zona, C. Modulation of AMPA Receptors in Cultured Cortical Neurons Induced by the Antiepileptic Drug Levetiracetam. Epilepsia 2007, 48, 654–662. [Google Scholar] [CrossRef]
- Wang, M.J.; Jiang, L.; Chen, H.S.; Cheng, L. Levetiracetam Protects Against Cognitive Impairment of Subthreshold Convulsant Discharge Model Rats by Activating Protein Kinase C (PKC)-Growth-Associated Protein 43 (GAP-43)-Calmodulin-Dependent Protein Kinase (CaMK) Signal Transduction Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 4627–4638. [Google Scholar] [CrossRef] [PubMed]
- Béïque, J.C.; Andrade, R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J. Physiol. 2003, 546, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef]
- Das, M.; Mao, W.; Shao, E.; Tamhankar, S.; Yu, G.Q.; Yu, X.; Ho, K.; Wang, X.; Wang, J.; Mucke, L. Interdependence of neural network dysfunction and microglial alterations in Alzheimer’s disease-related models. iScience 2021, 24, 103245. [Google Scholar] [CrossRef]
- Das, M.; Maeda, S.; Hu, B.; Yu, G.-Q.; Guo, W.; Lopez, I.; Yu, X.; Tai, C.; Wang, X.; Mucke, L. Neuronal levels and sequence of tau modulate the power of brain rhythms. Neurobiol. Dis. 2018, 117, 181–188. [Google Scholar] [CrossRef]
- Alavi, M.S.; Fanoudi, S.; Hosseini, M.; Sadeghnia, H.R. Beneficial effects of levetiracetam in streptozotocin-induced rat model of Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.Y.; Zhang, H.C.; Lv, Y.D.; Jin, F.Y.; Wu, X.J.; Zhu, J.; Ruan, Y. Levetiracetam alleviates cognitive decline in Alzheimer’s disease animal model by ameliorating the dysfunction of the neuronal network. Front. Aging Neurosci. 2022, 14, 888784. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011; p. 246. [Google Scholar]
- Dickey, C.; Kraft, C.; Jinwal, U.; Koren, J.; Johnson, A.; Anderson, L.; Lebson, L.; Lee, D.; Dickson, D.; De Silva, R.; et al. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am. J. Pathol. 2009, 174, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Onos, K.D.; Quinney, S.K.; Jones, D.R.; Masters, A.R.; Pandey, R.; Keezer, K.J.; Biesdorf, C.; Metzger, I.F.; Meyers, J.A.; Peters, J.; et al. Pharmacokinetic, pharmacodynamic, and transcriptomic analysis of chronic levetiracetam treatment in 5XFAD mice: A MODEL-AD preclinical testing core study. Alzheimer’s Dement. 2022, 8, e12329. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Baldauf, K.; Wetzel, W.; Reymann, K.G. Effects of methylphenidate on the behavior of male 5xFAD mice. Pharmacol. Biochem. Behav. 2015, 128, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, C.; Chaparro, R.E.; Karlnoski, R.; Erasso, D.; Gordon, M.; Morgan, D.; Bosco, G.; Rubini, A.; Parmagnani, A.; Paoli, A.; et al. Effects of repetitive exposure to anesthetics and analgesics in the Tg2576 mouse Alzheimer’s model. Neurotox. Res. 2014, 26, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Shi, R.; Li, V.; Wang, J.; Zhang, R.; Tempier, A.; He, J.; Kong, J.; Wang, J.F.; Li, X.M. Quetiapine Attenuates Glial Activation and Proinflammatory Cytokines in APP/PS1 Transgenic Mice via Inhibition of Nuclear Factor-κB Pathway. Int. J. Neuropsychopharmacol. 2015, 18, pyu022. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; He, J.; Zhang, R.; Kong, L.; Tempier, A.; Kong, J.; Li, X.-M. Therapeutic effects of quetiapine on memory deficit and brain β-amyloid plaque pathology in a transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 270–278. [Google Scholar] [CrossRef]
- Alamed, J.; Wilcock, D.M.; Diamond, D.M.; Gordon, M.N.; Morgan, D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc. 2006, 1, 1671–1679. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Davtyan, H.; Serraneau, K.; Jules, P.; Zitnyar, A.; Pressman, E.; Zagorski, K.; Antonyan, T.; Hovakimyan, A.; Paek, H.J.; et al. Active immunization with tau epitope in a mouse model of tauopathy induced strong antibody response together with improvement in short memory and pSer396-tau pathology. Neurobiol. Dis. 2020, 134, 104636. [Google Scholar] [CrossRef]
- Lee, D.C.; Rizer, J.; Selenica, M.-L.B.; Reid, P.; Kraft, C.; Johnson, A.; Blair, L.; Gordon, M.N.; Dickey, C.A.; Morgan, D.J. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflamm. 2010, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Bucuvalas, J.C.; Mazariegos, G.V.; Magee, J.C.; Sanchez-Fueyo, A.; Spain, K.M.; Lesniak, A.; Kanaparthi, S.; Perito, E.; Venkat, V.L.; et al. Efficacy and Safety of Immunosuppression Withdrawal in Pediatric Liver Transplant Recipients: Moving Toward Personalized Management. Hepatology 2021, 73, 1985–2004. [Google Scholar] [CrossRef]
- Isse, K.; Lesniak, A.; Grama, K.; Maier, J.; Specht, S.; Castillo-Rama, M.; Lunz, J.; Roysam, B.; Michalopoulos, G.; Demetris, A.J. Preexisting epithelial diversity in normal human livers: A tissue-tethered cytometric analysis in portal/periportal epithelial cells. Hepatology 2013, 57, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, M.L.; Joly-Amado, A.; Azam, S.; Elza, M.; Selenica, M.-L.; Pappas, C.; Small, B.; Engelman, R.; Gordon, M.N.; Morgan, D. Partial rescue of memory deficits induced by calorie restriction in a mouse model of tau deposition. Behav. Brain Res. 2014, 271, 79–88. [Google Scholar] [CrossRef]
- Gonçalves, J.; Bicker, J.; Gouveia, F.; Liberal, J.; Oliveira, R.C.; Alves, G.; Falcão, A.; Fortuna, A. Nose-to-brain delivery of levetiracetam after intranasal administration to mice. Int. J. Pharm. 2019, 564, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.J.; Hirsch, E.; Boehrer, A.; Noyer, M.; Marescaux, C. Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Res. 1995, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.; Albert, M.S.; Krauss, G.; Speck, C.L.; Gallagher, M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage. Clin. 2015, 7, 688–698. [Google Scholar] [CrossRef]
- Gillard, M.; Chatelain, P.; Fuks, B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur. J. Pharmacol. 2006, 536, 102–108. [Google Scholar] [CrossRef]
- Grizenko, N.; Cai, E.; Jolicoeur, C.; Ter-Stepanian, M.; Joober, R. Effects of methylphenidate on acute math performance in children with attention-deficit hyperactivity disorder. Can. J. Psychiatry. Rev. Can. De Psychiatr. 2013, 58, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.C.; Hunt, S.P.; Stanford, S.C. Behavioural and neurochemical abnormalities in mice lacking functional tachykinin-1 (NK1) receptors: A model of attention deficit hyperactivity disorder. Neuropharmacology 2009, 57, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Lanctot, K.L.; Chau, S.A.; Herrmann, N.; Drye, L.T.; Rosenberg, P.B.; Scherer, R.W.; Black, S.E.; Vaidya, V.; Bachman, D.L.; Mintzer, J.E. Effect of methylphenidate on attention in apathetic AD patients in a randomized, placebo-controlled trial. Int. Psychogeriatr. 2014, 26, 239–246. [Google Scholar] [CrossRef]
- Lanctôt, K.; Sankhe, K.; Perin, J.; Herrmann, N.; Brawman-Mintzer, O.; Lerner, A.J.; Mintzer, J.; Padala, P.R.; Rosenberg, P.B.; Shade, D.M.; et al. Correlation between changes in apathy and global cognition in apathetic associated with Alzheimer’s disease: Analysis of the Apathy Dementia Methylphenidate 2 (ADMET 2) clinical trial. Am. J. Geriatr. Psychiatry 2023, 31, S100–S101. [Google Scholar] [CrossRef]
- Gravielle, M.C. Activation-induced regulation of GABA receptors: Is there a link with the molecular basis of benzodiazepine tolerance? Pharmacol. Res. 2015, 109, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Liu, M.; He, J.; Guo, H.; Xue, M.; Wang, X.; Li, X.M. Quetiapine attenuates recognition memory impairment and hippocampal oxidative stress in a transgenic mouse model of Alzheimer’s disease. NeuroReport 2014, 25, 647–650. [Google Scholar] [CrossRef]
- Zhao, Z.; Luo, G.; Liu, M.; Guo, H.; Xue, M.; Wang, X.; Li, X.M.; He, J. Quetiapine reduces microglial number in the hippocampus of a transgenic mouse model of Alzheimer’s disease. NeuroReport 2014, 25, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.H.; Rodriguiz, R.M.; Caron, M.G.; Wetsel, W.C.; Rothman, R.B.; Roth, B.L. N-Desalkylquetiapine, a potent norepinephrine reuptake inhibitor and partial 5-HT1A agonist, as a putative mediator of quetiapine’s antidepressant activity. Neuropsychopharmacology 2008, 33, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Murakami, S.; Torigoe, N.; Kitamura, Y.; Asanuma, M. Neuroprotective effects of levetiracetam target xCT in astrocytes in parkinsonian mice. J. Neurochem. 2016, 136, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, 96, e52434. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Curzon, P.; Rustay, N.R.; Browman, K.E. Cued and Contextual Fear Conditioning for Rodents; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Victorino, D.B.; Pinheiro, D.; Scott-McKean, J.J.; Barker, S.; Stasko, M.R.; Faber, J.; Scorza, C.A.; Costa, A.C.S. Atypical electrophysiological and behavioral responses to diazepam in a leading mouse model of Down syndrome. Sci. Rep. 2021, 11, 9521. [Google Scholar] [CrossRef] [PubMed]
- Kinrys, G.; Worthington, J.J.; Wygant, L.; Nery, F.; Reese, H.; Pollack, M.H. Levetiracetam as adjunctive therapy for refractory anxiety disorders. J. Clin. Psychiatry 2007, 68, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Bartholome, O.; Van den Ackerveken, P.; Sánchez Gil, J.; de la Brassinne Bonardeaux, O.; Leprince, P.; Franzen, R.; Rogister, B. Puzzling Out Synaptic Vesicle 2 Family Members Functions. Front. Mol. Neurosci. 2017, 10, 148. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Vanaman, T.C.; Whiteheart, S.W.; Slevin, J.T. Levetiracetam prevents kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes. Epilepsia 2008, 49, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.R.; DeGulis, O.; Nomura, T.; Lee, S.; Hark, T.J.; Dynes, J.C.; Dexter, E.X.; Dulewicz, M.; Ge, J.; Upadhyay, A.; et al. Levetiracetam prevents Aβ(42) production through SV2a-dependent modulation of App processing in Alzheimer’s disease models. bioRxiv 2024. [Google Scholar] [CrossRef]
- Vossel, K.; Ranasinghe, K.G.; Beagle, A.J.; La, A.; Ah Pook, K.; Castro, M.; Mizuiri, D.; Honma, S.M.; Venkateswaran, N.; Koestler, M.; et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Sarkis, R.A. Levetiracetam in Alzheimer’s Disease: Do Epileptologists Already Have the Cure? Epilepsy Curr. 2022, 22, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Toniolo, S.; Tai, X.Y.; Akinola, M.; Symmonds, M.; Mura, S.; Galloway, J.; Hallam, A.; Chan, J.Y.C.; Koychev, I.; et al. Safety, tolerability and efficacy outcomes of the Investigation of Levetiracetam in Alzheimer’s Disease (ILiAD) trial. medRxiv 2024. [Google Scholar] [CrossRef]
- Belcastro, V.; Costa, C.; Galletti, F.; Pisani, F.; Calabresi, P.; Parnetti, L. Levetiracetam monotherapy in Alzheimer patients with late-onset seizures: A prospective observational study. Eur. J. Neurol. 2007, 14, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, C.S.; Baker, S.L.; Dobyns, L.; Janabi, M.; Jagust, W.J.; Harrison, T.M. Tau accumulation and atrophy predict amyloid independent cognitive decline in aging. Alzheimer’s Dement. 2024, 20, 2526–2537. [Google Scholar] [CrossRef]
- Huszár, Z.; Engh, M.A.; Pavlekovics, M.; Sato, T.; Steenkamp, Y.; Hanseeuw, B.; Terebessy, T.; Molnár, Z.; Hegyi, P.; Csukly, G. Risk of conversion to mild cognitive impairment or dementia among subjects with amyloid and tau pathology: A systematic review and meta-analysis. Alzheimer’s Res. Ther. 2024, 16, 81. [Google Scholar] [CrossRef]
- Mielke, M.M.; Frank, R.D.; Dage, J.L.; Jeromin, A.; Ashton, N.J.; Blennow, K.; Karikari, T.K.; Vanmechelen, E.; Zetterberg, H.; Algeciras-Schimnich, A.; et al. Comparison of Plasma Phosphorylated Tau Species With Amyloid and Tau Positron Emission Tomography, Neurodegeneration, Vascular Pathology, and Cognitive Outcomes. JAMA Neurol. 2021, 78, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Gidal, B.E.; Sheth, R.D.; Magnus, L.; Herbeuval, A.F. Levetiracetam does not alter body weight: Analysis of randomized, controlled clinical trials. Epilepsy Res. 2003, 56, 121–126. [Google Scholar] [CrossRef]
- Koul, R.; Al-Futaisi, A.M. Levetiracetam induced weight loss in two children. J. Pediatr. Neurol. 2008, 6, 257–260. [Google Scholar]
- Gelisse, P.; Juntas-Morales, R.; Genton, P.; Hillaire-Buys, D.; Diaz, O.; Coubes, P.; Crespel, A. Dramatic weight loss with levetiracetam. Epilepsia 2008, 49, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, W.O.; Lacey, A.S.; Thomas, R.H.; Smith, P.E.; Rees, M.I. Weight change associated with antiepileptic drugs. J. Neurol. Neurosurg. Psychiatry 2013, 84, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.A.; Shaikh, I.A.; Khateeb, M.M.; Habeeb, S.M. Omega 3 polyunsaturated fatty acids enhance the protective effect of levetiracetam against seizures, cognitive impairment and hippocampal oxidative DNA damage in young kindled rats. Pharmacol. Biochem. Behav. 2015, 135, 105–113. [Google Scholar] [CrossRef]
- Gillard, M.; Fuks, B.; Leclercq, K.; Matagne, A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: Relationship to anti-convulsant properties. Eur. J. Pharmacol. 2011, 664, 36–44. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation Parameter | Plasma | Brain |
|---|---|---|
| Calibration range (μg/mL) | 2.5–40 | 2.5–160 a |
| Regression equation b,c | y = 0.022641x – 0.013985 | y = 0.037154x – 0.013824 |
| Coefficient of determination (r2) d | 0.9948 | 0.9920 |
| LLOQ (μg/mL) | 2.5 | 0.625 |
| Inter-day | ||
| Precision (%CV) | 2.88–9.48 | 5.79–9.42 |
| Accuracy (%Bias) | −1.52–13.42 | −4.7–2.07 |
| Intra-day | ||
| Precision (%CV) | 2.81–9.91 | 2.17–6.33 |
| Accuracy (%Bias) | 0.60–8.88 | −7.61–4.45 |
| Recovery (%) | 58.86–79.18 | 69.10–87.07 |
| Ntg Chow | Tg4510 Chow | Tg4510 Dia | Tg4510 Lev | Tg4510 Mph | Tg4510 Quet | |
|---|---|---|---|---|---|---|
| BW (g) | 30.39 ± 0.58 | 27.475 ± 0.92 | 27.66 ± 1.01 | 28.17 ± 0.78 | 27.46 ± 0.62 | 29.60 ± 0.67 |
| Brain weight (mg) | 476.60 ± 2.23 | 392.17 ± 1.0 a | 392.33 ± 1.21 a | 392.83 ± 1.35 a | 393.40 ± 1.86 a | 392.20 ± 0.43 a |
| BAT (mg) | 149.80 ± 2.95 | 126.17 ± 2.94 | 138.33 ± 4.54 | 167.17 ± 4.99 | 143.80 ± 3.61 | 132.80 ± 2.75 |
| Total muscle (mg) | 282.40 ± 1.60 | 270.50 ± 1.76 | 275.67 ± 2.54 | 280.67 ± 1.68 | 255.60 ± 2.74 | 278.00 ± 1.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, J.C.; Levis Rabi, M.; Novoa, M.; Nash, K.R.; Joly-Amado, A. Evaluating the Efficacy of Levetiracetam on Non-Cognitive Symptoms and Pathology in a Tau Mouse Model. Biomedicines 2024, 12, 2891. https://doi.org/10.3390/biomedicines12122891
Thompson JC, Levis Rabi M, Novoa M, Nash KR, Joly-Amado A. Evaluating the Efficacy of Levetiracetam on Non-Cognitive Symptoms and Pathology in a Tau Mouse Model. Biomedicines. 2024; 12(12):2891. https://doi.org/10.3390/biomedicines12122891
Chicago/Turabian StyleThompson, Jackson C., Marselina Levis Rabi, Michelle Novoa, Kevin R. Nash, and Aurelie Joly-Amado. 2024. "Evaluating the Efficacy of Levetiracetam on Non-Cognitive Symptoms and Pathology in a Tau Mouse Model" Biomedicines 12, no. 12: 2891. https://doi.org/10.3390/biomedicines12122891
APA StyleThompson, J. C., Levis Rabi, M., Novoa, M., Nash, K. R., & Joly-Amado, A. (2024). Evaluating the Efficacy of Levetiracetam on Non-Cognitive Symptoms and Pathology in a Tau Mouse Model. Biomedicines, 12(12), 2891. https://doi.org/10.3390/biomedicines12122891