
Sodium Phenylbutyrate Attenuates Cisplatin-Induced Acute Kidney Injury Through Inhibition of Pyruvate Dehydrogenase Kinase 4

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Histological and Immunohistochemical Analyses
2.3. Serum Analysis
2.4. TUNEL Analysis
2.5. Cell Culture and Western Blot Analysis
2.6. Mitochondrial Reactive Oxygen Species and Membrane Potential Measurement
2.7. Mitochondrial Oxygen Consumption Rate Measurement
2.8. Quantitative and Semi-Quantitative PCR
2.9. Cell Survival Assay
2.10. Statistical Analysis
3. Results
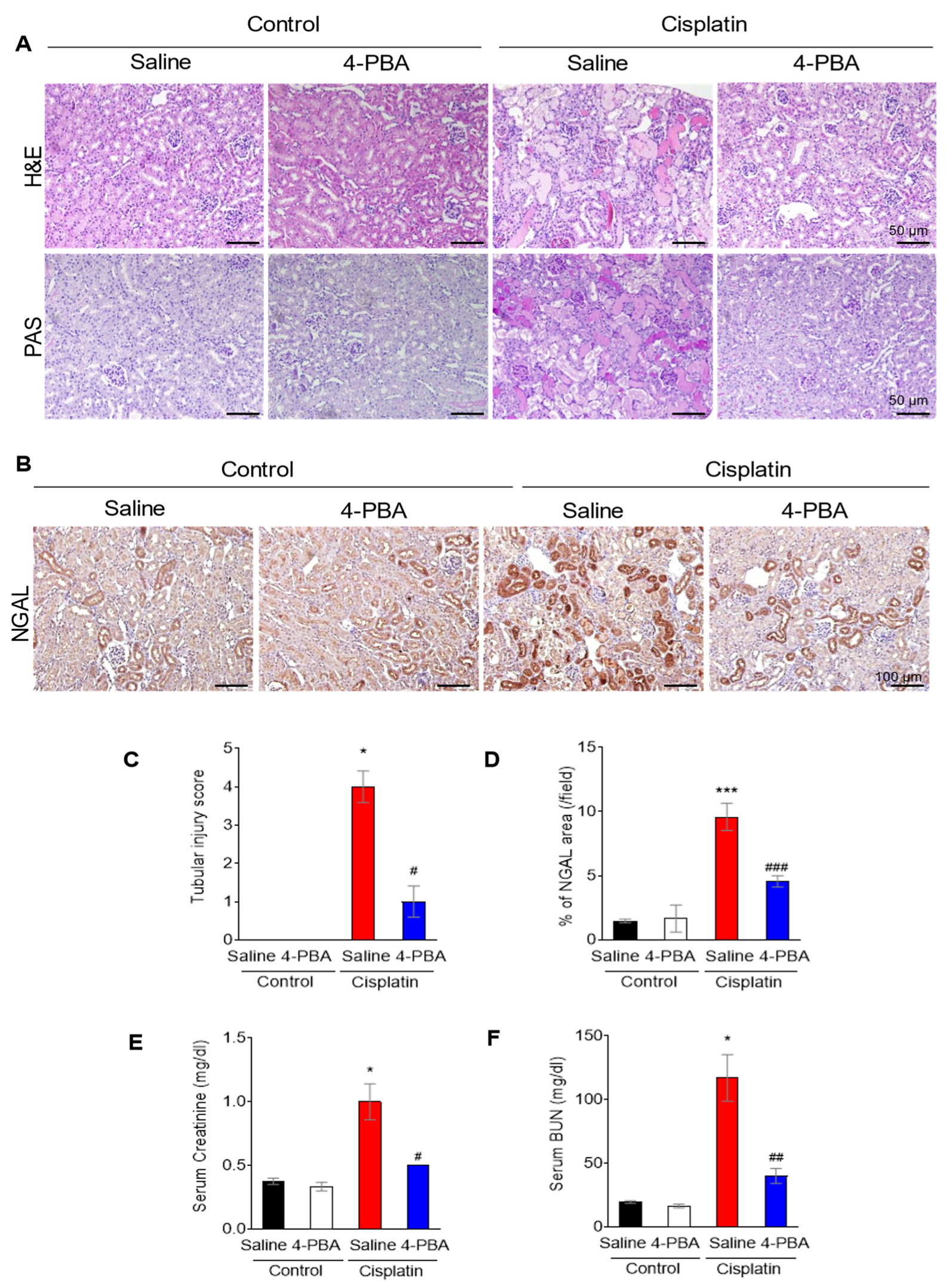
3.1. 4-PBA Attenuates Cisplatin-Induced Acute Kidney Injury
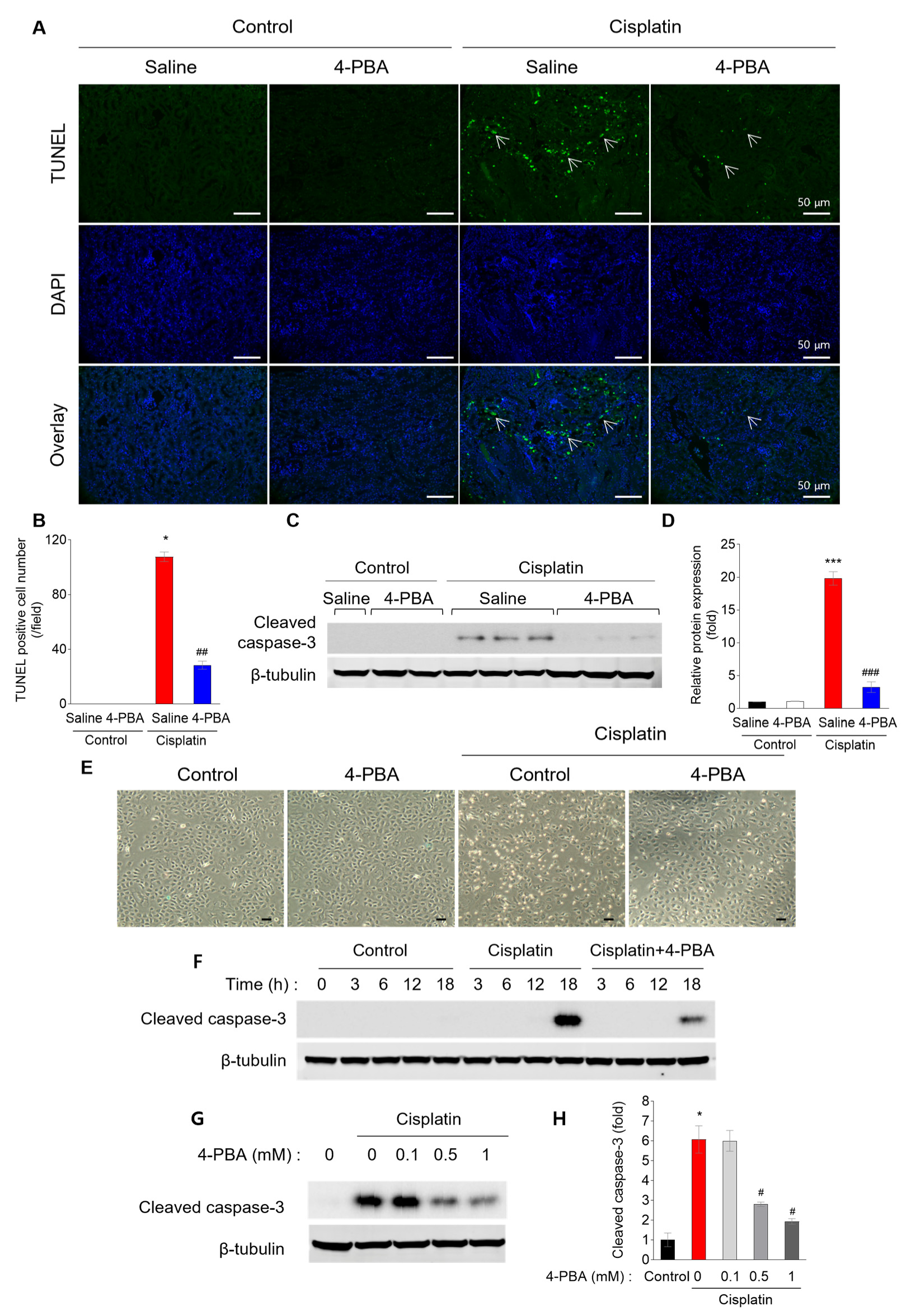
3.2. 4-PBA Prevents Cisplatin-Induced Renal Tubular Apoptosis
3.3. 4-PBA Attenuates Cisplatin-Induced Inflammation
3.4. 4-PBA Attenuates Cisplatin-Induced Reactive Oxygen Species and Mitochondrial Damage
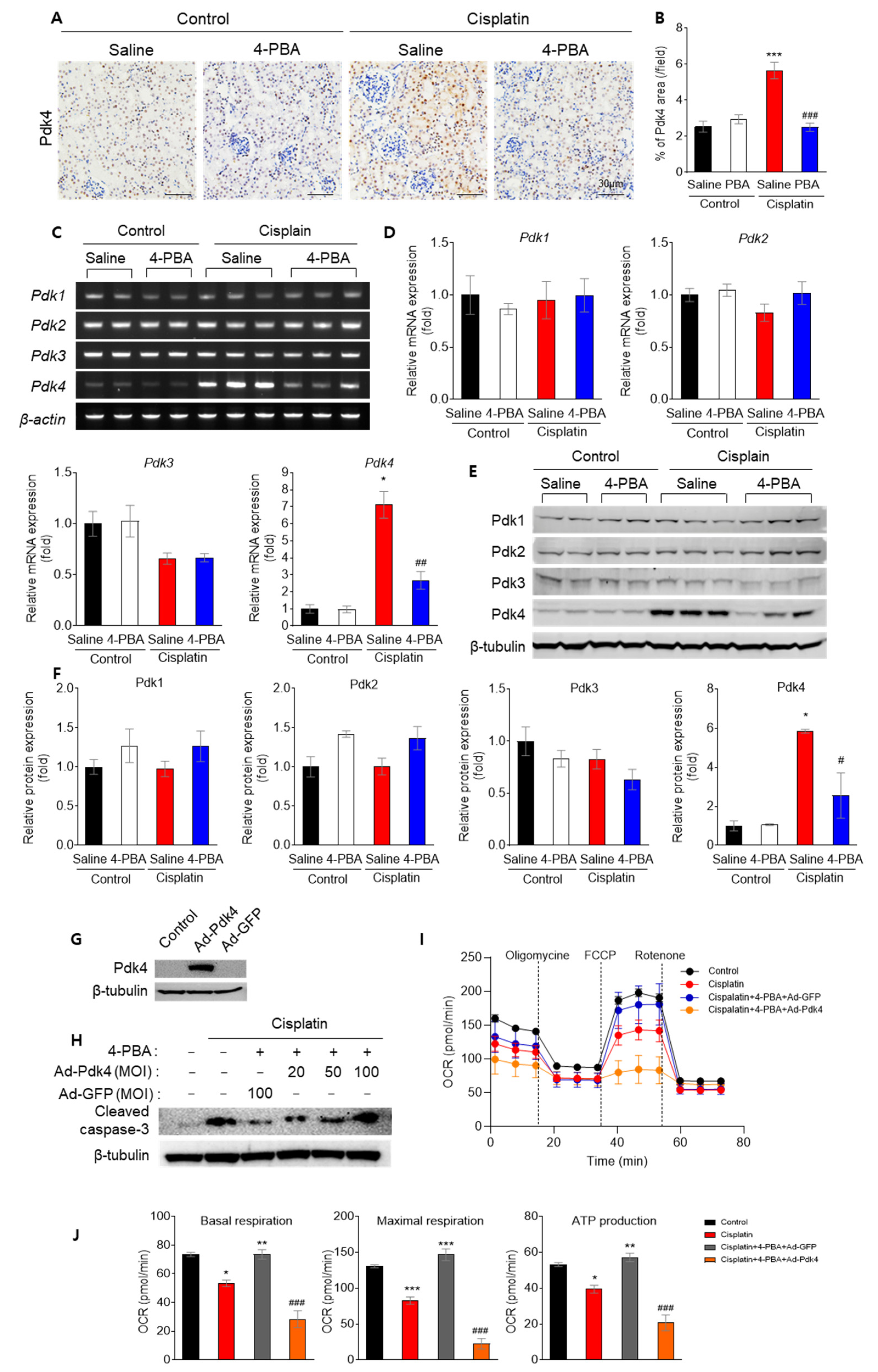
3.5. 4-PBA Diminishes Apoptosis and Mitochondrial Dysfunction by Reducing Pdk4
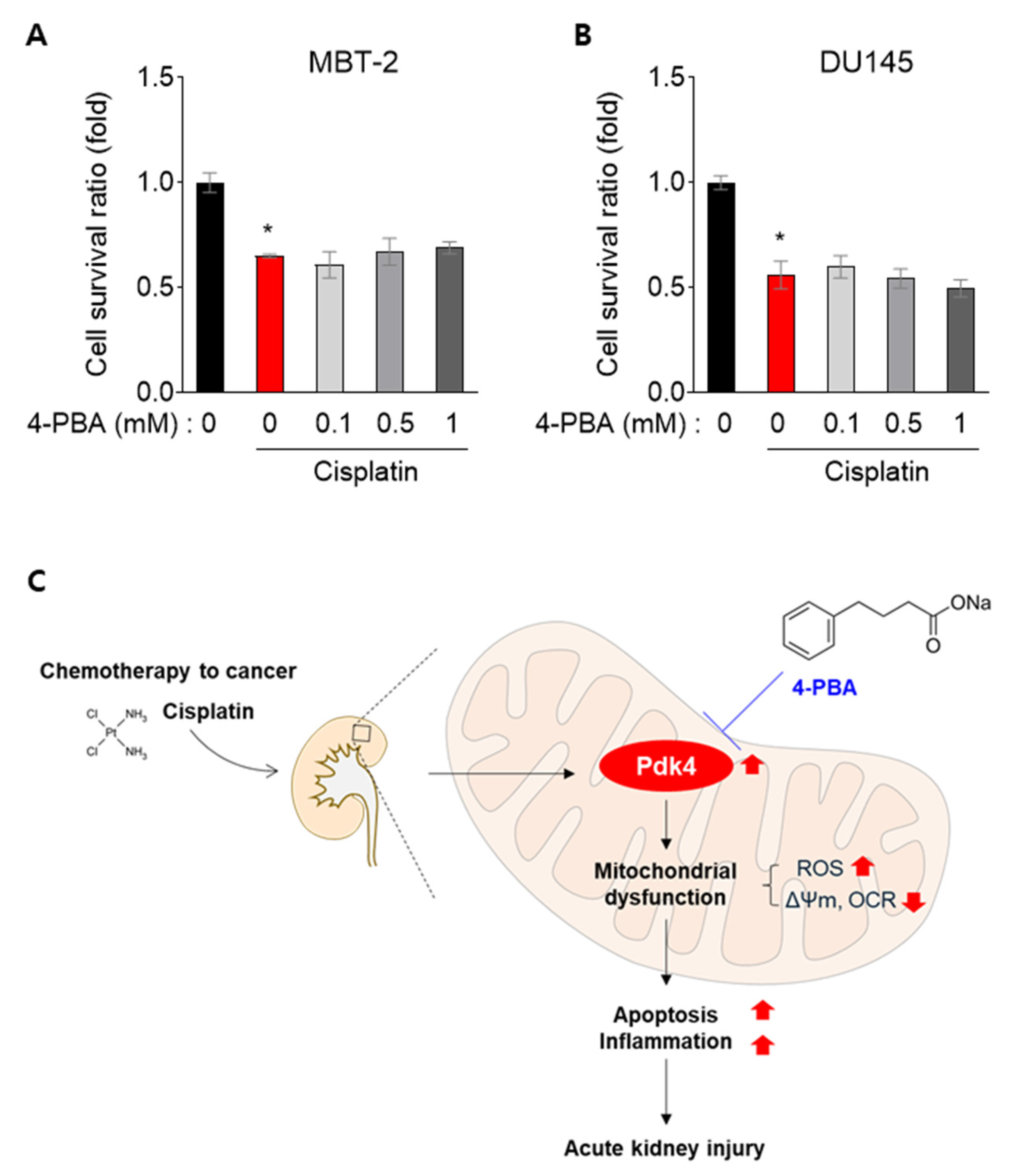
3.6. 4-PBA Does Not Interfere with the Anti-Cancer Effect in MBT-2 and DU145 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H.; Liu, F.; Dong, Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch. Toxicol. 2014, 88, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef]
- Oh, C.J.; Ha, C.M.; Choi, Y.K.; Park, S.; Choe, M.S.; Jeoung, N.H.; Huh, Y.H.; Kim, H.J.; Kweon, H.S.; Lee, J.M.; et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017, 91, 880–895. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Y.; Zhou, W.; Wu, Y.; Zhang, S.; Ding, G.; Zhang, Y.; Zhang, A.; Huang, S.; Jia, Z.; et al. Maintaining homeostasis of mitochondria and endoplasmic reticulum with NSC228155 alleviates cisplatin-induced acute kidney injury. Free Radic. Biol. Med. 2022, 181, 270–287. [Google Scholar] [CrossRef]
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Carlisle, R.E.; Brimble, E.; Werner, K.E.; Cruz, G.L.; Ask, K.; Ingram, A.J.; Dickhout, J.G. 4-Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PLoS ONE 2014, 9, e84663. [Google Scholar] [CrossRef]
- Ferriero, R.; Iannuzzi, C.; Manco, G.; Brunetti-Pierri, N. Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. J. Inherit. Metab. Dis. 2015, 38, 895–904. [Google Scholar] [CrossRef]
- Kim, M.J.; Sinam, I.S.; Siddique, Z.; Jeon, J.H.; Lee, I.K. The Link between Mitochondrial Dysfunction and Sarcopenia: An Update Focusing on the Role of Pyruvate Dehydrogenase Kinase 4. Diabetes Metab. J. 2023, 47, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Wang, H.; Cui, N. mTOR pathway mediates endoplasmic reticulum stress-induced CD4(+) T cell apoptosis in septic mice. Apoptosis 2022, 27, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, D.C.; Zhang, J.; Hsu, S.W.; Weiss, R.H.; Chen, C.H. A Novel Renoprotective Strategy: Upregulation of PD-L1 Mitigates Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2021, 22, 13304. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mesentier-Louro, L.A.; Oh, A.J.; Heng, K.; Shariati, M.A.; Huang, H.; Hu, Y.; Liao, Y.J. Increased ER Stress After Experimental Ischemic Optic Neuropathy and Improved RGC and Oligodendrocyte Survival After Treatment with Chemical Chaperon. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1953–1966. [Google Scholar] [CrossRef]
- Oh, G.S.; Kim, H.J.; Choi, J.H.; Shen, A.; Choe, S.K.; Karna, A.; Lee, S.H.; Jo, H.J.; Yang, S.H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKα-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef]
- Li, Y.M.; Li, Y.; Yan, L.; Wang, H.; Wu, X.J.; Tang, J.T.; Wang, L.L.; Shi, Y.Y. Comparison of urine and blood NGAL for early prediction of delayed graft function in adult kidney transplant recipients: A meta-analysis of observational studies. BMC Nephrol. 2019, 20, 291. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Conway, E.M.; Harris, R.C. Survivin mediates renal proximal tubule recovery from AKI. J. Am. Soc. Nephrol. 2013, 24, 2023–2033. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-α. Kidney Int. 2004, 65, 490–499. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Flaig, T.W.; Spiess, P.E.; Abern, M.; Agarwal, N.; Bangs, R.; Buyyounouski, M.K.; Chan, K.; Chang, S.S.; Chang, P.; Friedlander, T.; et al. NCCN Guidelines(R) Insights: Bladder Cancer, Version 3.2024. J. Natl. Compr. Cancer Netw. 2024, 22, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, K.; Ljungman, M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer 2008, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Al-Keilani, M.S.; Alzoubi, K.H.; Jaradat, S.A. The effect of combined treatment with sodium phenylbutyrate and cisplatin, erlotinib, or gefitinib on resistant NSCLC cells. Clin. Pharmacol. 2018, 10, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, R.; Manco, G.; Lamantea, E.; Nusco, E.; Ferrante, M.I.; Sordino, P.; Stacpoole, P.W.; Lee, B.; Zeviani, M.; Brunetti-Pierri, N. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci. Transl. Med. 2013, 5, 175ra131. [Google Scholar] [CrossRef]
- Oh, C.J.; Kim, M.J.; Lee, J.M.; Kim, D.H.; Kim, I.Y.; Park, S.; Kim, Y.; Lee, K.B.; Lee, S.H.; Lim, C.W.; et al. Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion. Kidney Int. 2023, 104, 724–739. [Google Scholar] [CrossRef]
- Lee, K.-M.; Hwang, Y.J.; Jung, G.-S. Alantolactone Attenuates Renal Fibrosis via Inhibition of Transforming Growth Factor β/Smad3 Signaling Pathway. Diabetes Metab. J. 2024, 48, 72–82. [Google Scholar] [CrossRef]
- Thoudam, T.; Chanda, D.; Sinam, I.S.; Kim, B.G.; Kim, M.J.; Oh, C.J.; Lee, J.Y.; Kim, M.J.; Park, S.Y.; Lee, S.Y.; et al. Noncanonical PDK4 action alters mitochondrial dynamics to affect the cellular respiratory status. Proc. Natl. Acad. Sci. USA 2022, 119, e2120157119. [Google Scholar] [CrossRef]
- Thoudam, T.; Chanda, D.; Lee, J.Y.; Jung, M.-K.; Sinam, I.S.; Kim, B.-G.; Park, B.-Y.; Kwon, W.H.; Kim, H.-J.; Kim, M.; et al. Enhanced Ca2+-channeling complex formation at the ER-mitochondria interface underlies the pathogenesis of alcohol-associated liver disease. Nat. Commun. 2023, 14, 1703. [Google Scholar] [CrossRef]
- Khang, A.R.; Kim, D.H.; Kim, M.-J.; Oh, C.J.; Jeon, J.-H.; Choi, S.H.; Lee, I.-K. Reducing Oxidative Stress and Inflammation by Pyruvate Dehydrogenase Kinase 4 Inhibition Is Important in Prevention of Renal Ischemia-Reperfusion Injury in Diabetic Mice. Diabetes Metab. J. 2024, 48, 405–417. [Google Scholar] [CrossRef]
- Francescato, H.D.; Costa, R.S.; Junior, F.B.; Coimbra, T.M. Effect of JNK inhibition on cisplatin-induced renal damage. Nephrol. Dial. Transplant. 2007, 22, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Wang, H.; Zhu, J.; Fu, Y.; Cai, J.; Chen, A.; Tang, C.; Dong, Z. Endoplasmic reticulum stress contributes to cisplatin-induced chronic kidney disease via the PERK-PKCdelta pathway. Cell. Mol. Life Sci. 2022, 79, 452. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xie, B.; Mao, Z. Autophagy in premature senescent cells is activated via AMPK pathway. Int. J. Mol. Sci. 2012, 13, 3563–3582. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Mattos, S.; Villalonga, P.; Clardy, J.; Lam, E.W. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol. Cancer Ther. 2008, 7, 3237–3246. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Su, J.; Xie, Q.; Ma, L.; Zeng, L.; Yu, Y.; Liu, S.; Li, S.; Li, Z.; et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol. Rep. 2015, 34, 3051–3060. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, C.J.; Choi, W.; Lee, H.Y.; Lee, I.-K.; Kim, M.-J.; Jeon, J.-H. Sodium Phenylbutyrate Attenuates Cisplatin-Induced Acute Kidney Injury Through Inhibition of Pyruvate Dehydrogenase Kinase 4. Biomedicines 2024, 12, 2815. https://doi.org/10.3390/biomedicines12122815
Oh CJ, Choi W, Lee HY, Lee I-K, Kim M-J, Jeon J-H. Sodium Phenylbutyrate Attenuates Cisplatin-Induced Acute Kidney Injury Through Inhibition of Pyruvate Dehydrogenase Kinase 4. Biomedicines. 2024; 12(12):2815. https://doi.org/10.3390/biomedicines12122815
Chicago/Turabian StyleOh, Chang Joo, Wooyoung Choi, Ha Young Lee, In-Kyu Lee, Min-Ji Kim, and Jae-Han Jeon. 2024. "Sodium Phenylbutyrate Attenuates Cisplatin-Induced Acute Kidney Injury Through Inhibition of Pyruvate Dehydrogenase Kinase 4" Biomedicines 12, no. 12: 2815. https://doi.org/10.3390/biomedicines12122815
APA StyleOh, C. J., Choi, W., Lee, H. Y., Lee, I.-K., Kim, M.-J., & Jeon, J.-H. (2024). Sodium Phenylbutyrate Attenuates Cisplatin-Induced Acute Kidney Injury Through Inhibition of Pyruvate Dehydrogenase Kinase 4. Biomedicines, 12(12), 2815. https://doi.org/10.3390/biomedicines12122815