
Tau Protein and β-Amyloid Associated with Neurodegeneration in Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis (EAE), a Mouse Model of Multiple Sclerosis


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. EAE-Inducted Mice
2.3. Evaluation of EAE and Phases of EAE Disease
2.4. Sample Collection
2.5. Histopathological Staining
2.6. Immunohistochemical Staining
2.7. Morphometry
2.8. Image Collection and Statistical Analysis
3. Results
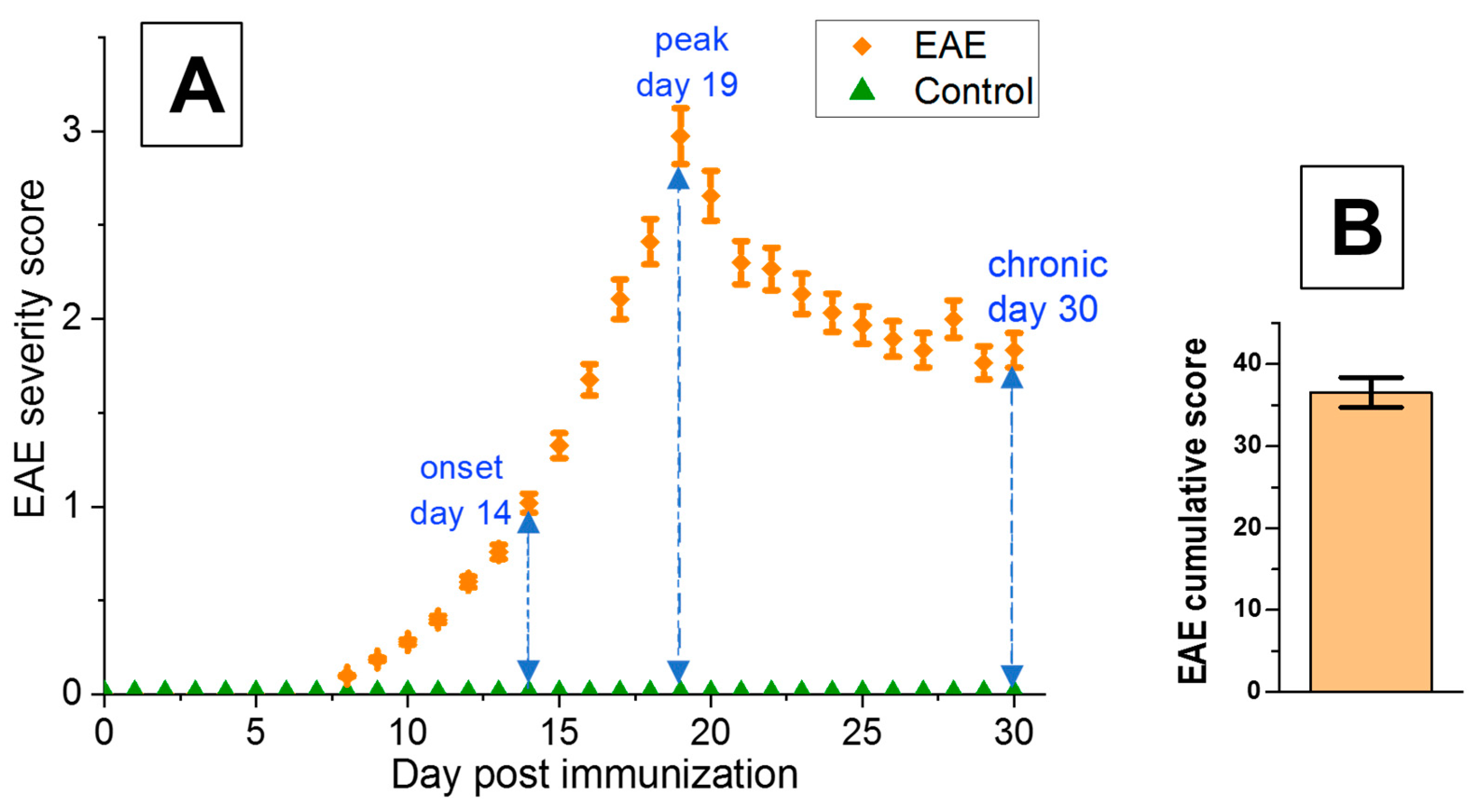
3.1. Characterization of EAE Severity
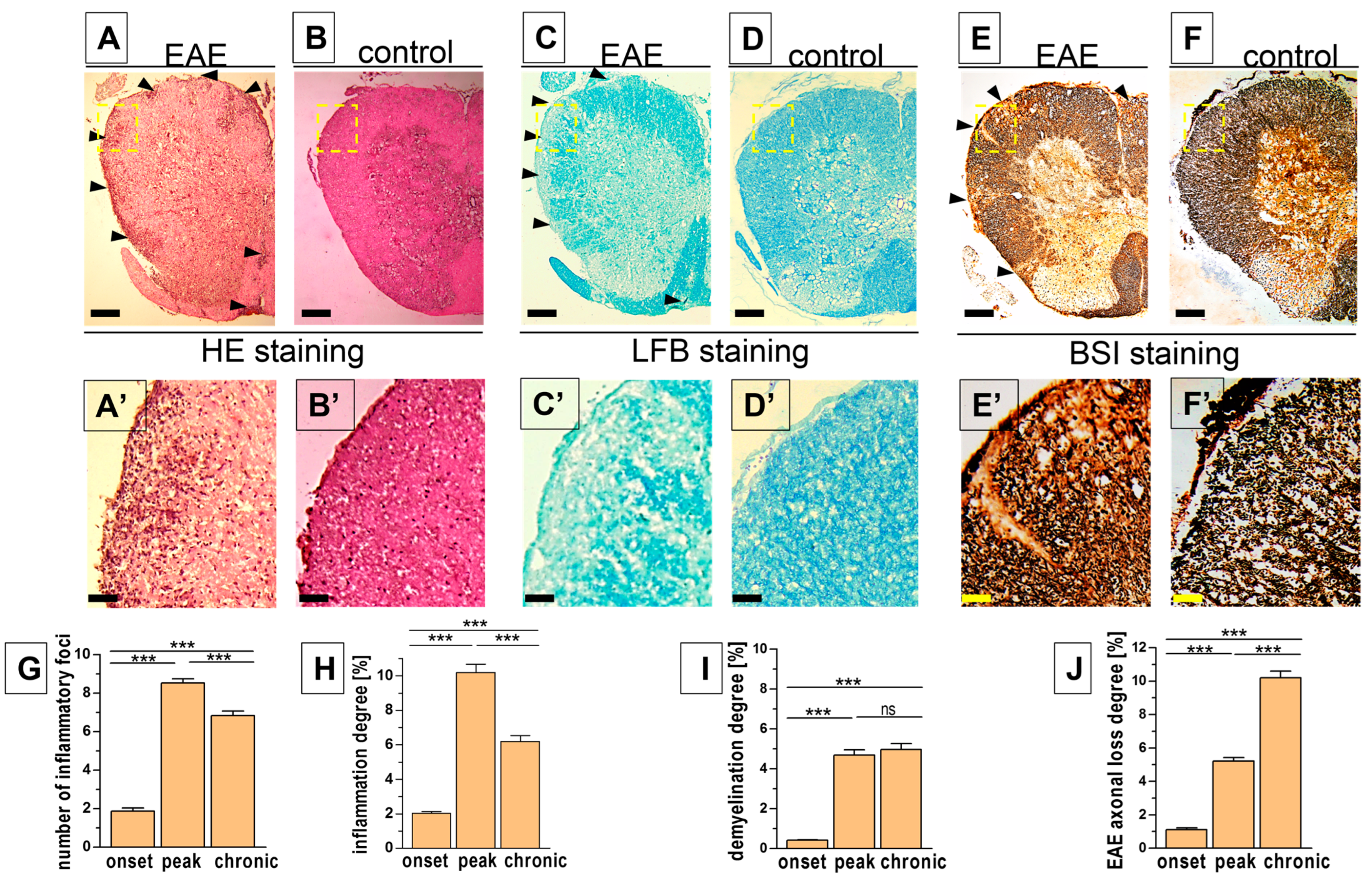
3.2. Histopathological Changes in the Spinal Cord
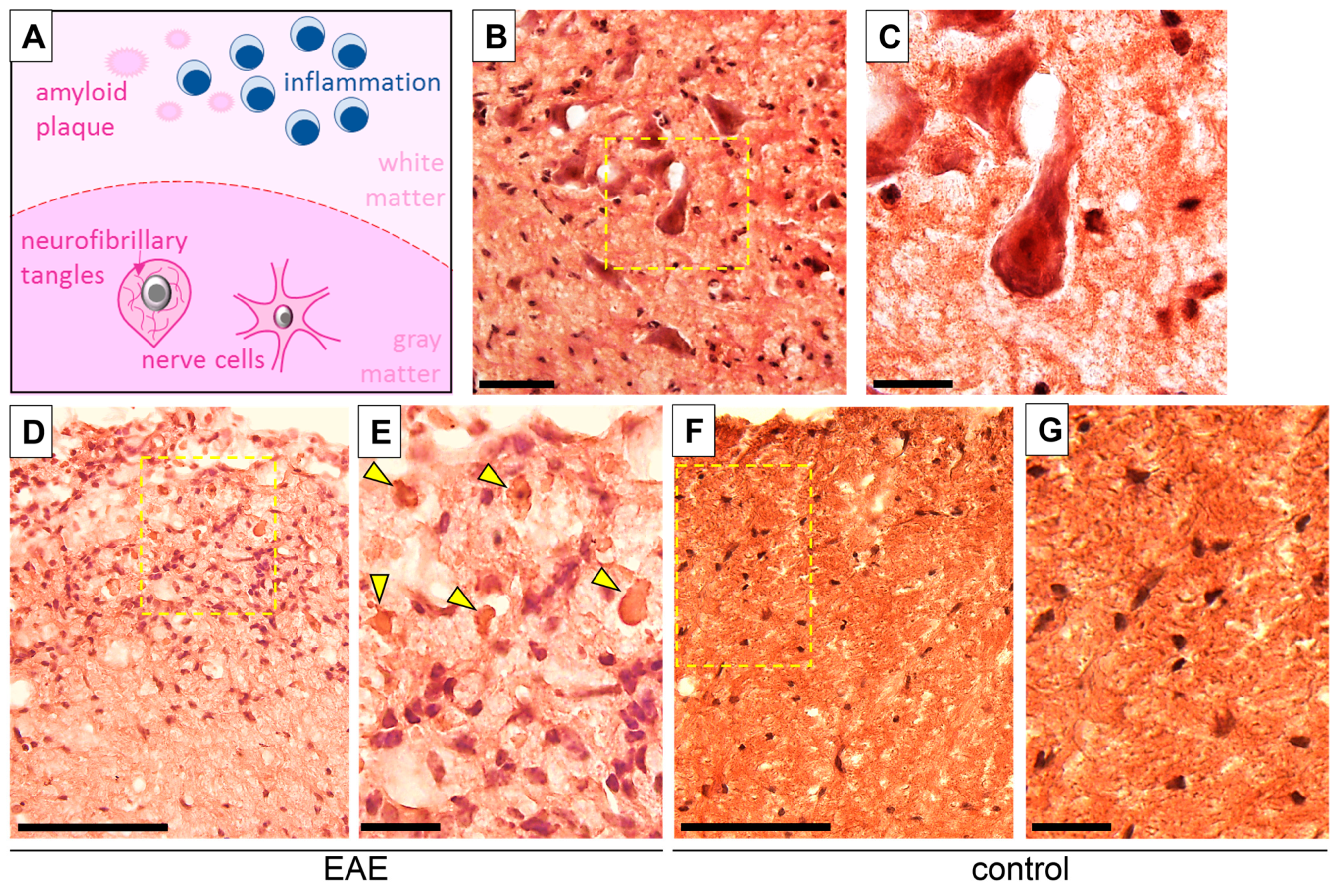
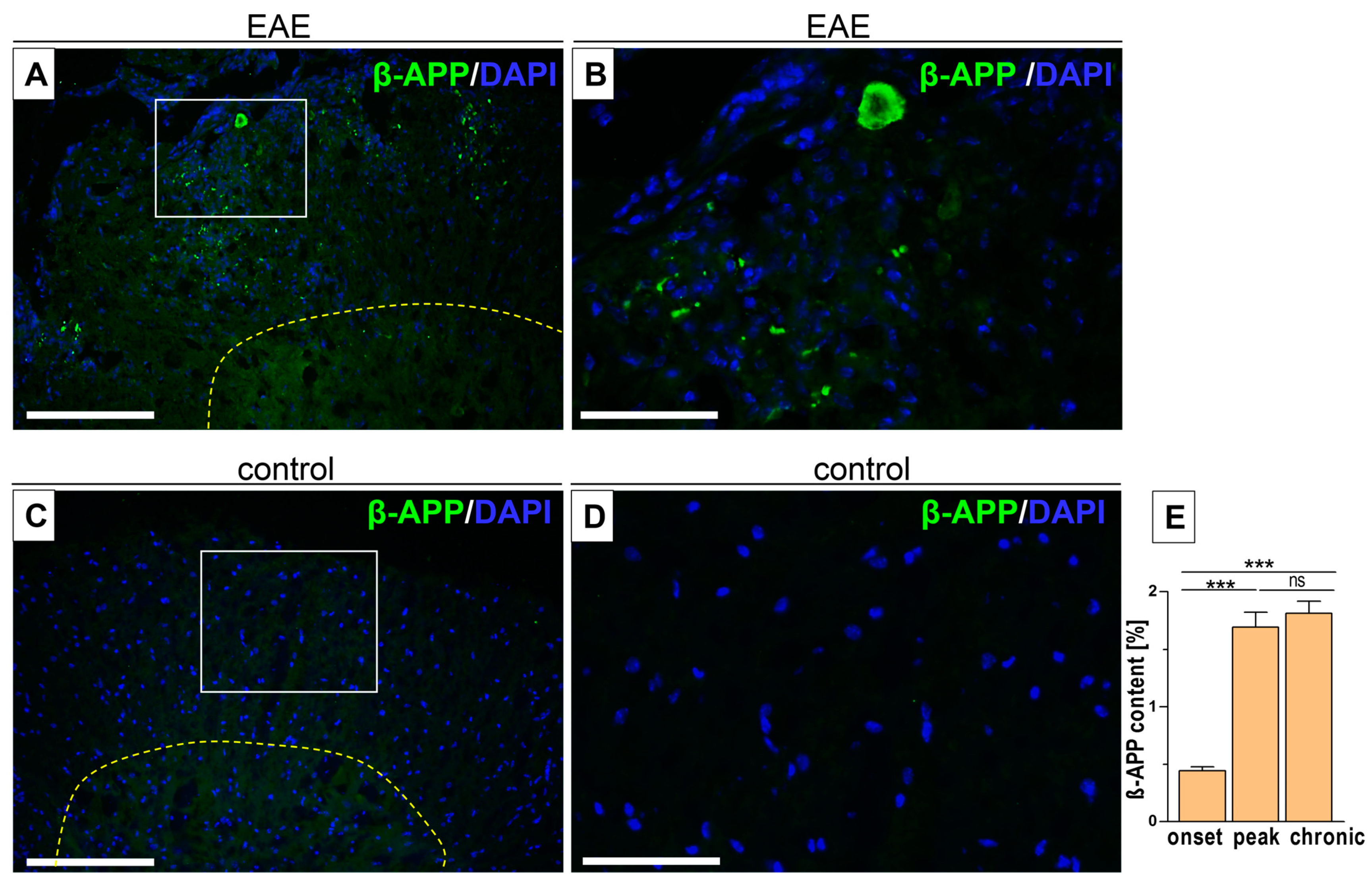
3.3. Intercellular β-APP Aggregation and Expression Level
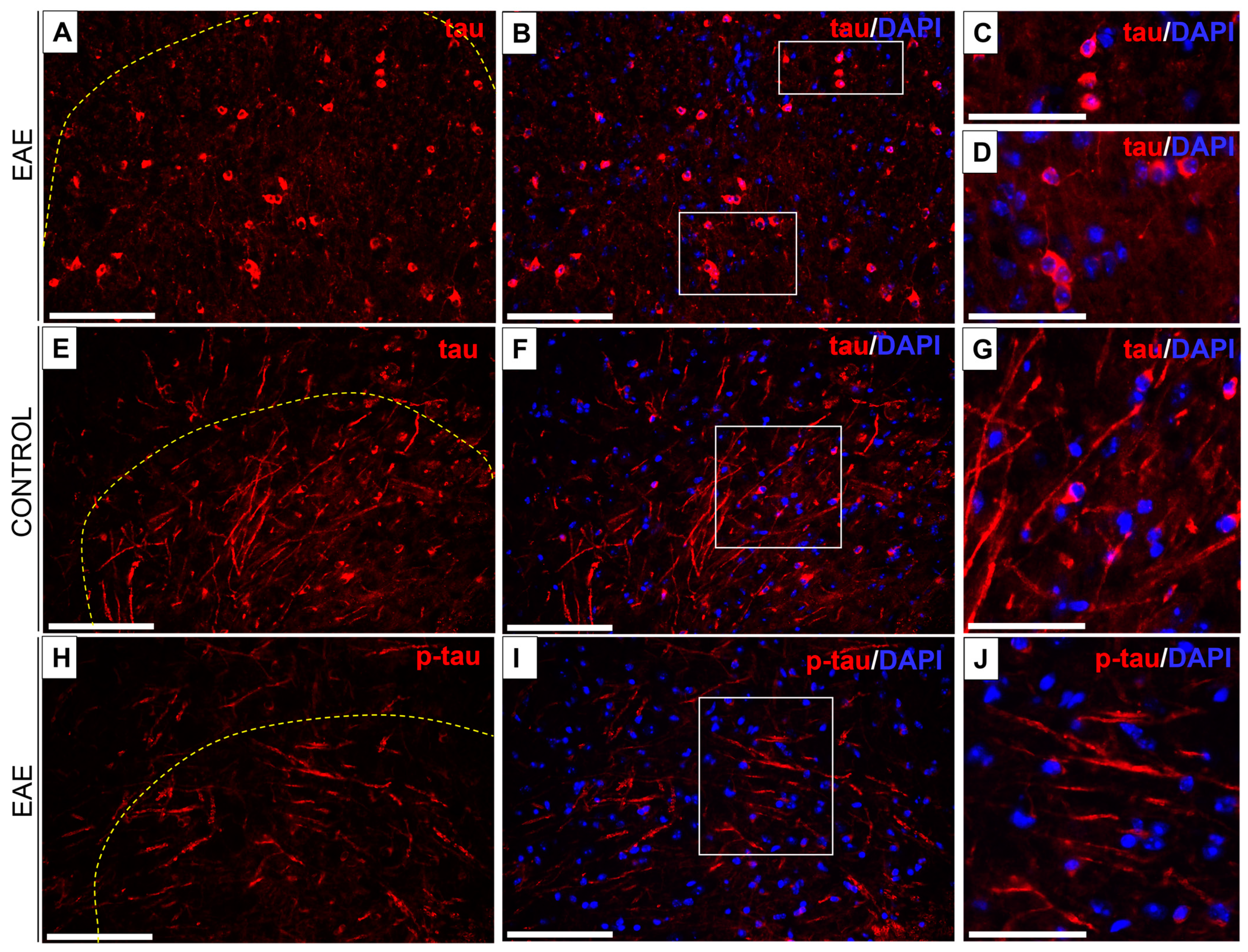
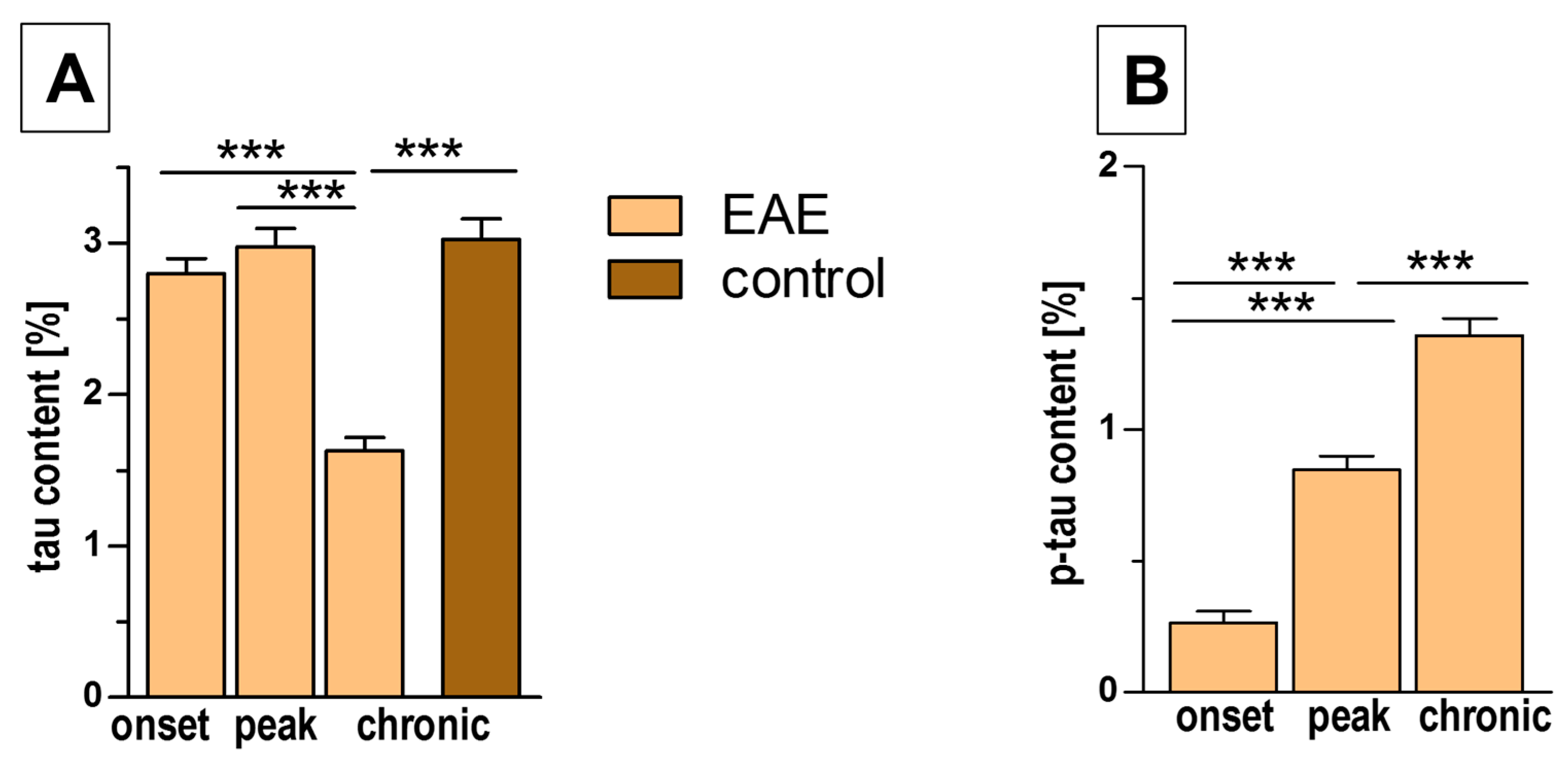
3.4. Tau/Phospho-Tau (p-Tau) Expression in EAE and Control Mice
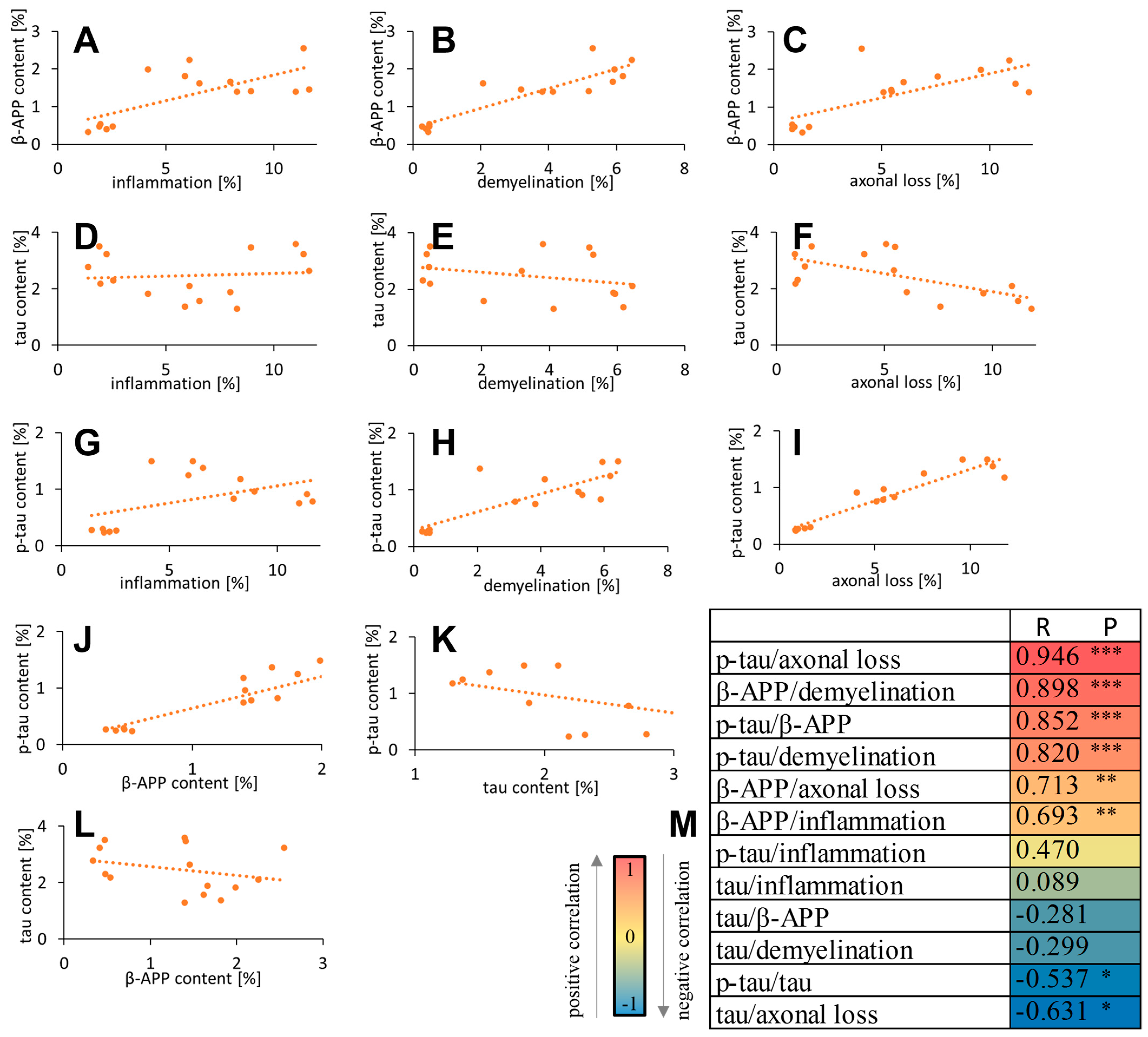
3.5. Correlation of β-APP/Tau/p-Tau with Inflammation and Neurodegeneration Parameters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baker, D.; Gerritsen, W.; Rundle, J.; Amor, S. Critical appraisal of animal models of multiple sclerosis. Mult. Scler. 2011, 17, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Ffrench-Constant, C. Pathogenesis of multiple sclerosis. Lancet 1994, 343, 271–275. [Google Scholar] [CrossRef]
- Haque, A.; Trager, N.N.M.; Butler, J.T.; Das, A.; Zaman, V.; Banik, N.L. A novel combination approach to effectively reduce inflammation and neurodegeneration in multiple sclerosis models. Neurochem. Int. 2024, 175, 105697. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. Serum-Based Biomarkers in Neurodegeneration and Multiple Sclerosis. Biomedicines 2022, 10, 1077. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-beta and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V. Beta-Amyloid precursor protein (APP) and the human diseases. AIMS Neurosci. 2019, 6, 273–281. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef]
- Ousman, S.S.; Tomooka, B.H.; van Noort, J.M.; Wawrousek, E.F.; O’Conner, K.; Hafler, D.A.; Sobel, R.A.; Robinson, W.H.; Steinman, L. Protective and therapeutic role for alpha B-crystallin in autoimmune demyelination. Nature 2007, 448, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.L.; Ghosn, E.E.; Axtell, R.C.; Herges, K.; Kuipers, H.F.; Woodling, N.S.; Andreasson, K.; Herzenberg, L.A.; Herzenberg, L.A.; Steinman, L. Reversal of paralysis and reduced inflammation from peripheral administration of beta-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med. 2012, 4, 145ra105. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Banati, R.B.; Cuzner, M.L.; Kreutzberg, G.W.; Newcombe, J. Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia 1995, 15, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A. Role of amyloid from a multiple sclerosis perspective: A literature review. Neuroimmunomodulation 2015, 22, 343–346. [Google Scholar] [CrossRef]
- Pyka-Fosciak, G.; Fosciak, M.; Wojcik, B.; Lis, G.J.; Litwin, J.A. Histopatological parameters of the spinal cord in different phases of experimental autoimmune encephalomyelitis. A mouse model of multiple sclerosis examined by classical stainings combined with immunohistochemistry. J. Physiol. Pharmacol. 2023, 74, 465–472. [Google Scholar]
- Dapson, R.W. Amyloid from a histochemical perspective. A review of the structure, properties and types of amyloid, and a proposed staining mechanism for Congo red staining. Biotech. Histochem. 2018, 93, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Pyka-Fosciak, G.; Stasiolek, M.; Litwin, J.A. Immunohistochemical analysis of spinal cord components in mouse model of experimental autoimmune encephalomyelitis. Folia Histochem. Cytobiol 2018, 56, 151–158. [Google Scholar] [CrossRef]
- Weigand, A.J.; Bangen, K.J.; Thomas, K.R.; Delano-Wood, L.; Gilbert, P.E.; Brickman, A.M.; Bondi, M.W. Is tau in the absence of amyloid on the Alzheimer’s continuum?: A study of discordant PET positivity; Alzheimer’s Disease Neuroimaging Initiative. Brain Commun. 2020, 2, fcz046. [Google Scholar] [CrossRef]
- Pyka-Fościak, G.; Zemła, J.; Lis, G.J.; Litwin, J.A.; Lekka, M. Changes in spinal cord stiffness in the course of experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Arch. Biochem. Biophys. 2020, 15, 108221. [Google Scholar] [CrossRef]
- Pyka-Fościak, G.; Lis, G.J.; Litwin, J.A. Adhesion molecule profile and the effect of anti-VLA-4 mAb treatment in experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Int. J. Mol. Sci. 2022, 23, 4637. [Google Scholar] [CrossRef]
- Pyka-Fościak, G.; Fościak, M.; Pabijan, J.; Lis, G.J.; Litwin, J.A.; Lekka, M. Changes in stiffness of the optic nerve and involvement of neurofilament light chains in the course of experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166796. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenarative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121. [Google Scholar] [CrossRef]
- Weinger, J.G.; Davies, P.; Acker, C.M.; Brosnan, C.F.; Tsiperson, V.; Bayewitz, A.; Shafit-Zagardo, B.J. Neuropathol. Mice devoid of Tau have increased susceptibility to neuronal damage in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. Exp. Neurol. 2012, 71, 422–433. [Google Scholar]
- Anderson, J.; Hampton, D.; Patani, R.; Pryce, G.; Crowther, R.; Reynolds, R.; Franklin, R.J.M.; Giovannoni, G.; Compston, D.A.S.; Baker, D.; et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain 2008, 131, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- LaCroix, M.S.; Mirbaha, H.; Shang, P.; Zandee, S.; Foong, C.; Prat, A.; White, C.L., 3rd; Stuve, O.; Diamond, M.I. Tau seeding in cases of multiple sclerosis. Acta Neuropathol. Commun. 2022, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef] [PubMed]
- Cras, P.; Kawai, M.; Siedlak, S.; Perry, G. Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res. 1991, 558, 312–314. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 26, 856376. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 16, 815347. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef]
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Brück, W. Acute axonal injury in multiple sclerosis: Correlation with demyelination and inflammation. Brain 2000, 123, 1174–1183. [Google Scholar] [PubMed]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 2023, 618, 349–357. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EAE Mice | Onset Phase | Peak Phase | Chronic Phase |
|---|---|---|---|
| day post-immunization | 14 | 19 | 30 |
| mean EAE score | 1.02 ± 0.05 | 2.97 ± 0.15 | 1.83 ± 0.09 |
| cumulative score | - | - | 36.52 ± 1.83 |
| number of inflammatory foci per cross-section | 1.88 ± 0.17 | 8.52 ± 0.22 | 6.84 ± 0.23 |
| inflammation area [%] | 2.02 ± 0.11 | 10.19 ± 0.48 | 6.20 ± 0.33 |
| demyelination area [%] | 0.41 ± 0.03 | 4.67 ± 0.28 | 4.95 ± 0.30 |
| axonal loss area [%] | 1.12 ± 0.11 | 5.21 ± 0.21 | 10.21 ± 0.38 |
| EAE | Control | |||
|---|---|---|---|---|
| Onset Phase | Peak Phase | Chronic Phase | ||
| β-amyloid content [%] | 0.44 ± 0.03 | 1.69 ± 0.13 | 1.81 ± 0.11 | - |
| tau content [%] | 2.80 ± 0.10 | 2.98 ± 0.12 | 1.63 ± 0.09 | 3.02 ± 0.14 |
| p-tau content [%] | 0.26 ± 0.04 | 0.85 ± 0.05 | 1.36 ± 0.07 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyka-Fościak, G.; Jasek-Gajda, E.; Wójcik, B.; Lis, G.J.; Litwin, J.A. Tau Protein and β-Amyloid Associated with Neurodegeneration in Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis (EAE), a Mouse Model of Multiple Sclerosis. Biomedicines 2024, 12, 2770. https://doi.org/10.3390/biomedicines12122770
Pyka-Fościak G, Jasek-Gajda E, Wójcik B, Lis GJ, Litwin JA. Tau Protein and β-Amyloid Associated with Neurodegeneration in Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis (EAE), a Mouse Model of Multiple Sclerosis. Biomedicines. 2024; 12(12):2770. https://doi.org/10.3390/biomedicines12122770
Chicago/Turabian StylePyka-Fościak, Grażyna, Ewa Jasek-Gajda, Bożena Wójcik, Grzegorz J. Lis, and Jan A. Litwin. 2024. "Tau Protein and β-Amyloid Associated with Neurodegeneration in Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis (EAE), a Mouse Model of Multiple Sclerosis" Biomedicines 12, no. 12: 2770. https://doi.org/10.3390/biomedicines12122770
APA StylePyka-Fościak, G., Jasek-Gajda, E., Wójcik, B., Lis, G. J., & Litwin, J. A. (2024). Tau Protein and β-Amyloid Associated with Neurodegeneration in Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis (EAE), a Mouse Model of Multiple Sclerosis. Biomedicines, 12(12), 2770. https://doi.org/10.3390/biomedicines12122770