
Current Therapeutic Opportunities for Estrogen Receptor Mutant Breast Cancer

Abstract
1. Introduction
2. ERα Domain Structure
3. ERα in Human Breast Cancer
3.1. Endocrine Therapy in ER+ Breast Cancer
3.2. Endocrine-Resistant Breast Cancer
3.3. ESR1 Mutation
3.4. Therapeutic Strategies for ESR1 Mutant Breast Cancer
4. SERDs in Clinical Trials
4.1. Giredestrant (GDC-9545)
4.2. Imlunestrant (LY3484356)
4.3. Camizestrant (AZD-9833)
4.4. Vepdegestrant (ARV-471)
4.5. Palazestrant (OP-1250)
5. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar] [CrossRef] [PubMed]
- Toft, D.; Gorski, J. A receptor molecule for estrogens: Isolation from the rat uterus and preliminary characterization. Proc. Natl. Acad. Sci. USA 1966, 55, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to verb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef]
- Siersbaek, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor alpha function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef]
- Brisken, C.; O’Malley, B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010, 2, a003178. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Manka, D.; Wagner, K.U.; Khan, S.A. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 14718–14723. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Palaniappan, M.; Nguyen, L.; Grimm, S.L.; Xi, Y.; Xia, Z.; Li, W.; Coarfa, C. The genomic landscape of estrogen receptor alpha binding sites in mouse mammary gland. PLoS ONE 2019, 14, e0220311. [Google Scholar] [CrossRef] [PubMed]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Gosden, J.R.; Middleton, P.G.; Rout, D. Localization of the human oestrogen receptor gene to chromosome 6q24–q27 by in situ hybridization. Cytogenet. Cell Genet. 1986, 43, 218–220. [Google Scholar] [CrossRef]
- Green, S.; Kumar, V.; Krust, A.; Walter, P.; Chambon, P. Structural and functional domains of the estrogen receptor. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 2, 751–758. [Google Scholar] [CrossRef]
- Sarwar, N.; Kim, J.S.; Jiang, J.; Peston, D.; Sinnett, H.D.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr. Relat. Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Finn, G.; Solodin, N.M.; Singarapu, K.K.; Sahu, S.C.; Markley, J.L.; Kadunc, K.J.; Ellison-Zelski, S.J.; Kariagina, A.; Haslam, S.Z.; et al. Regulation of estrogen receptor alpha N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol. Cell Biol. 2012, 32, 445–457. [Google Scholar] [CrossRef]
- Klein-Hitpass, L.; Ryffel, G.U.; Heitlinger, E.; Cato, A.C. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 1988, 16, 647–663. [Google Scholar] [CrossRef]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Pestell, R.; Curran, E.M.; Welshons, W.V.; Fuqua, S.A. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004, 64, 9199–9208. [Google Scholar] [CrossRef] [PubMed]
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008, 22, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Korach, K.S. The physiological role of estrogen receptor functional domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O’Malley, B.W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 2000, 5, 939–948. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. Molecular Pathways: Targeting Steroid Receptor Coactivators in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5403–5407. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef]
- Nichols, M.; Rientjes, J.M.; Stewart, A.F. Different positioning of the ligand-binding domain helix 12 and the F domain of the estrogen receptor accounts for functional differences between agonists and antagonists. EMBO J. 1998, 17, 765–773. [Google Scholar] [CrossRef]
- Schwartz, J.A.; Zhong, L.; Deighton-Collins, S.; Zhao, C.; Skafar, D.F. Mutations targeted to a predicted helix in the extreme carboxyl-terminal region of the human estrogen receptor-alpha alter its response to estradiol and 4-hydroxytamoxifen. J. Biol. Chem. 2002, 277, 13202–13209. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Zhao, C.; Naganuma, M.; Abrams, J.; Deighton-Collins, S.; Skafar, D.F.; Koide, S. Identification of regions within the F domain of the human estrogen receptor alpha that are important for modulating transactivation and protein-protein interactions. Mol. Endocrinol. 2007, 21, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Korach, K.S. The F domain of estrogen receptor alpha is involved in species-specific, tamoxifen-mediated transactivation. J. Biol. Chem. 2018, 293, 8495–8507. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef]
- Jagannathan, V.; Robinson-Rechavi, M. Meta-analysis of estrogen response in MCF-7 distinguishes early target genes involved in signaling and cell proliferation from later target genes involved in cell cycle and DNA repair. BMC Syst. Biol. 2011, 5, 138. [Google Scholar] [CrossRef]
- Welboren, W.J.; Sweep, F.C.; Span, P.N.; Stunnenberg, H.G. Genomic actions of estrogen receptor alpha: What are the targets and how are they regulated? Endocr. Relat. Cancer 2009, 16, 1073–1089. [Google Scholar] [CrossRef]
- Palaniappan, M.; Edwards, D.; Creighton, C.J.; Medina, D.; Conneely, O.M. Reprogramming of the estrogen responsive transcriptome contributes to tamoxifen-dependent protection against tumorigenesis in the p53 null mammary epithelial cells. PLoS ONE 2018, 13, e0194913. [Google Scholar] [CrossRef]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Klein, P.; Tiersten, A.; Sparano, J.A. An emerging generation of endocrine therapies in breast cancer: A clinical perspective. NPJ Breast Cancer 2023, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Rej, R.K.; Roy, J.; Allu, S.R. Therapies for the Treatment of Advanced/Metastatic Estrogen Receptor-Positive Breast Cancer: Current Situation and Future Directions. Cancers 2024, 16, 552. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.R.; Jhaveri, K.; Kalinsky, K.; Bardia, A.; Wander, S.A. Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer. Nat. Rev. Clin. Oncol. 2024, 21, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr. Relat. Cancer 2014, 21, R235–R246. [Google Scholar] [CrossRef]
- Cole, M.P.; Jones, C.T.; Todd, I.D. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br. J. Cancer 1971, 25, 270–275. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Buschmann, M.; Wiegand, A.; Schnellbacher, K.; Bonn, R.; Rehe, A.; Trenk, D.; Jahnchen, E.; Roskamm, H. Comparison of the effects of two different galenical preparations of glyceryl trinitrate on pulmonary artery pressure and on the finger pulse curve. Eur. J. Clin. Pharmacol. 1993, 44, 451–456. [Google Scholar] [CrossRef]
- Nabholtz, J.M. Long-term safety of aromatase inhibitors in the treatment of breast cancer. Ther. Clin. Risk Manag. 2008, 4, 189–204. [Google Scholar] [CrossRef]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90 (Suppl. S1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.W. The history and mechanism of action of fulvestrant. Clin. Breast Cancer 2005, 6 (Suppl. S1), S5–S8. [Google Scholar] [CrossRef] [PubMed]
- Wardley, A.M. Fulvestrant: A review of its development, pre-clinical and clinical data. Int. J. Clin. Pract. 2002, 56, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, S.; Simigdala, N.; Ribas, R.; Schuster, E.; Leal, M.F.; Nikitorowicz-Buniak, J.; Rega, C.; Bihani, T.; Patel, H.; Johnston, S.R.; et al. Elacestrant demonstrates strong anti-estrogenic activity in PDX models of estrogen-receptor positive endocrine-resistant and fulvestrant-resistant breast cancer. NPJ Breast Cancer 2022, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.; Nicholson, R.I.; Bundred, N.J.; Anderson, E.; Rayter, Z.; Dowsett, M.; Fox, J.N.; Gee, J.M.; Webster, A.; Wakeling, A.E.; et al. Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer Res. 2001, 61, 6739–6746. [Google Scholar]
- Hoy, S.M. Elacestrant: First Approval. Drugs 2023, 83, 555–561. [Google Scholar] [CrossRef]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), Has Antitumor Activity in Multiple ER(+) Breast Cancer Patient-derived Xenograft Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4793–4804. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Jager, A.; de Vries, E.G.E.; der Houven van Oordt, C.W.M.; Neven, P.; Venema, C.M.; Glaudemans, A.; Wang, Y.; Bagley, R.G.; Conlan, M.G.; Aftimos, P. A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor availability and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using (18)F-FES PET/CT imaging. Breast Cancer Res. BCR 2020, 22, 97. [Google Scholar] [CrossRef]
- Garner, F.; Shomali, M.; Paquin, D.; Lyttle, C.R.; Hattersley, G. RAD1901: A novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 2015, 26, 948–956. [Google Scholar] [CrossRef]
- Patel, H.K.; Tao, N.; Lee, K.M.; Huerta, M.; Arlt, H.; Mullarkey, T.; Troy, S.; Arteaga, C.L.; Bihani, T. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. BCR 2019, 21, 146. [Google Scholar] [CrossRef] [PubMed]
- Gombos, A. Selective oestrogen receptor degraders in breast cancer: A review and perspectives. Curr. Opin. Oncol. 2019, 31, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Greene, G.L. Next-Generation ERalpha Inhibitors for Endocrine-Resistant ER+ Breast Cancer. Endocrinology 2019, 160, 759–769. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Wardell, S.E. The molecular mechanisms underlying the pharmacological actions of ER modulators: Implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol. 2010, 10, 620–628. [Google Scholar] [CrossRef]
- McDonnell, D.P. The molecular pharmacology of estrogen receptor modulators: Implications for the treatment of breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 871s–877s. [Google Scholar] [CrossRef]
- Puhalla, S.; Bhattacharya, S.; Davidson, N.E. Hormonal therapy in breast cancer: A model disease for the personalization of cancer care. Mol. Oncol. 2012, 6, 222–236. [Google Scholar] [CrossRef]
- Guglielmi, G.; Del Re, M.; Gol, L.S.; Bengala, C.; Danesi, R.; Fogli, S. Pharmacological insights on novel oral selective estrogen receptor degraders in breast cancer. Eur. J. Pharmacol. 2024, 969, 176424. [Google Scholar] [CrossRef]
- Nardone, A.; De Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The changing role of ER in endocrine resistance. Breast 2015, 24 (Suppl. S2), S60–S66. [Google Scholar] [CrossRef]
- Will, M.; Liang, J.; Metcalfe, C.; Chandarlapaty, S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nat. Rev. Cancer 2023, 23, 673–685. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Morrison, G.; Fu, X.; Shea, M.; Nanda, S.; Giuliano, M.; Wang, T.; Klinowska, T.; Osborne, C.K.; Rimawi, M.F.; Schiff, R. Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast Cancer Res. Treat. 2014, 144, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Joo, H.S.; Won, H.Y.; Min, K.W.; Kim, H.Y.; Son, T.; Oh, Y.H.; Lee, J.Y.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Agboke, F.A.; Cunliffe, H.E.; Ramos, P.; Jordan, V.C. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur. J. Cancer 2014, 50, 2866–2876. [Google Scholar] [CrossRef]
- Jeffreys, S.A.; Powter, B.; Balakrishnar, B.; Mok, K.; Soon, P.; Franken, A.; Neubauer, H.; de Souza, P.; Becker, T.M. Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells 2020, 9, 2077. [Google Scholar] [CrossRef]
- Gururaj, A.E.; Rayala, S.K.; Vadlamudi, R.K.; Kumar, R. Novel mechanisms of resistance to endocrine therapy: Genomic and nongenomic considerations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1001s–1007s. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar] [PubMed]
- Oesterreich, S.; Davidson, N.E. The search for ESR1 mutations in breast cancer. Nat. Genet. 2013, 45, 1415–1416. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J.A. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. Metab. TEM 2015, 26, 467–476. [Google Scholar] [CrossRef]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Carlson, K.E.; Choi, I.; Gee, A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: Evidence that an open pocket conformation is required for ligand interaction. Biochemistry 1997, 36, 14897–14905. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Zundelevich, A.; Dadiani, M.; Kahana-Edwin, S.; Itay, A.; Sella, T.; Gadot, M.; Cesarkas, K.; Farage-Barhom, S.; Saar, E.G.; Eyal, E.; et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res. BCR 2020, 22, 16. [Google Scholar] [CrossRef]
- Hancock, G.R.; Gertz, J.; Jeselsohn, R.; Fanning, S.W. Estrogen Receptor Alpha Mutations, Truncations, Heterodimers, and Therapies. Endocrinology 2024, 165, bqae051. [Google Scholar] [CrossRef] [PubMed]
- Dustin, D.; Gu, G.; Beyer, A.R.; Herzog, S.K.; Edwards, D.G.; Lin, H.; Gonzalez, T.L.; Grimm, S.L.; Coarfa, C.; Chan, D.W.; et al. RON signalling promotes therapeutic resistance in ESR1 mutant breast cancer. Br. J. Cancer 2021, 124, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, Y.; Yates, M.E.; Tasdemir, N.; Bahreini, A.; Chen, J.; Levine, K.M.; Priedigkeit, N.M.; Nasrazadani, A.; Ali, S.; et al. Hotspot ESR1 Mutations Are Multimodal and Contextual Modulators of Breast Cancer Metastasis. Cancer Res. 2022, 82, 1321–1339. [Google Scholar] [CrossRef] [PubMed]
- Grinshpun, A.; Sandusky, Z.M.; Jeselsohn, R. The Clinical Utility of ESR1 Mutations in Hormone Receptor-Positive, HER2-Negative Advanced Breast Cancer. Hematol. Oncol. Clin. N. Am. 2023, 37, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Grinshpun, A.; Chen, V.; Sandusky, Z.M.; Fanning, S.W.; Jeselsohn, R. ESR1 activating mutations: From structure to clinical application. Biochim. Et Biophys. Acta Rev. Cancer 2023, 1878, 188830. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef]
- Arnesen, S.; Blanchard, Z.; Williams, M.M.; Berrett, K.C.; Li, Z.; Oesterreich, S.; Richer, J.K.; Gertz, J. Estrogen Receptor Alpha Mutations in Breast Cancer Cells Cause Gene Expression Changes through Constant Activity and Secondary Effects. Cancer Res. 2021, 81, 539–551. [Google Scholar] [CrossRef]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; Nowak, J.; Sharma, S.K.; Hahm, J.B.; Kulp, K.; Hochberg, R.B.; Zhou, H.; Katzenellenbogen, J.A.; et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat. Chem. Biol. 2008, 4, 241–247. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, 12792. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Norris, J.D.; Chang, C.Y. Neomorphic ERalpha Mutations Drive Progression in Breast Cancer and Present a Challenge for New Drug Discovery. Cancer Cell 2018, 33, 153–155. [Google Scholar] [CrossRef]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.L.; Somerfield, M.R.; Dayao, Z.; Elias, A.; Kalinsky, K.; McShane, L.M.; Moy, B.; Park, B.H.; Shanahan, K.M.; Sharma, P.; et al. Biomarkers for Systemic Therapy in Metastatic Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. 2022, 40, 3205–3221. [Google Scholar] [CrossRef] [PubMed]
- Bellward, G.D.; Norstrom, R.J.; Whitehead, P.E.; Elliott, J.E.; Bandiera, S.M.; Dworschak, C.; Chang, T.; Forbes, S.; Cadario, B.; Hart, L.E.; et al. Comparison of polychlorinated dibenzodioxin levels with hepatic mixed-function oxidase induction in great blue herons. J. Toxicol. Environ. Health 1990, 30, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Wang, P.; Bahreini, A.; Gyanchandani, R.; Lucas, P.C.; Hartmaier, R.J.; Watters, R.J.; Jonnalagadda, A.R.; Trejo Bittar, H.E.; Berg, A.; Hamilton, R.L.; et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1130–1137. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidants and the central nervous system: Some fundamental questions. Is oxidant damage relevant to Parkinson’s disease, Alzheimer’s disease, traumatic injury or stroke? Acta Neurol. Scand. Suppl. 1989, 126, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Keenan, J.C.; Medford, A.J.; Dai, C.S.; Wander, S.A.; Spring, L.M.; Bardia, A. Novel oral selective estrogen receptor degraders (SERDs) to target hormone receptor positive breast cancer: Elacestrant as the poster-child. Expert Rev. Anticancer Ther. 2024, 24, 397–405. [Google Scholar] [CrossRef]
- Bardia, A.; Cortes, J.; Bidard, F.C.; Neven, P.; Garcia-Saenz, J.; Aftimos, P.; O’Shaughnessy, J.; Lu, J.; Tonini, G.; Scartoni, S.; et al. Elacestrant in ER+, HER2− Metastatic Breast Cancer with ESR1-Mutated Tumors: Subgroup Analyses from the Phase III EMERALD Trial by Prior Duration of Endocrine Therapy plus CDK4/6 Inhibitor and in Clinical Subgroups. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 4299–4309. [Google Scholar] [CrossRef]
- Bhatia, N.; Thareja, S. Elacestrant: A new FDA-approved SERD for the treatment of breast cancer. Med. Oncol. 2023, 40, 180. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Bellet, M.; Turner, N.C.; Loi, S.; Bardia, A.; Boni, V.; Sohn, J.; Neilan, T.G.; Villanueva-Vazquez, R.; Kabos, P.; et al. Phase Ia/b Study of Giredestrant +/- Palbociclib and +/- Luteinizing Hormone-Releasing Hormone Agonists in Estrogen Receptor-Positive, HER2-Negative, Locally Advanced/Metastatic Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zbieg, J.R.; Blake, R.A.; Chang, J.H.; Daly, S.; DiPasquale, A.G.; Friedman, L.S.; Gelzleichter, T.; Gill, M.; Giltnane, J.M.; et al. GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. J. Med. Chem. 2021, 64, 11841–11856. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Lim, E.; Chavez-MacGregor, M.; Bardia, A.; Wu, J.; Zhang, Q.; Nowecki, Z.; Cruz, F.M.; Safin, R.; Kim, S.B.; et al. Giredestrant for Estrogen Receptor-Positive, HER2-Negative, Previously Treated Advanced Breast Cancer: Results From the Randomized, Phase II acelERA Breast Cancer Study. J. Clin. Oncol. 2024, 42, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.L.; Lim, E.; Jeselsohn, R.; Ma, C.X.; Hamilton, E.P.; Osborne, C.; Bhave, M.; Kaufman, P.A.; Beck, J.T.; Manso Sanchez, L.; et al. Imlunestrant, an Oral Selective Estrogen Receptor Degrader, as Monotherapy and in Combination With Targeted Therapy in Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Phase Ia/Ib EMBER Study. J. Clin. Oncol. 2024, JCO2302733. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Zhao, B.; Shen, W.; Mur, C.; Barr, R.; Kindler, L.J.; Rubio, A.; Bastian, J.A.; Cohen, J.D.; Mattioni, B.E.; et al. Preclinical characterization of LY3484356, a novel, potent and orally bioavailable selective estrogen receptor degrader (SERD). Cancer Res. 2021, 81, 1236. [Google Scholar] [CrossRef]
- Hamilton, E.; Oliveira, M.; Turner, N.; Garcia-Corbacho, J.; Hernando, C.; Ciruelos, E.M.; Kabos, P.; Ruiz-Borrego, M.; Armstrong, A.; Patel, M.R.; et al. A phase I dose escalation and expansion trial of the next-generation oral SERD camizestrant in women with ER-positive, HER2-negative advanced breast cancer: SERENA-1 monotherapy results. Ann. Oncol. 2024, 35, 707–717. [Google Scholar] [CrossRef]
- Turner, N.; Huang-Bartlett, C.; Kalinsky, K.; Cristofanilli, M.; Bianchini, G.; Chia, S.; Iwata, H.; Janni, W.; Ma, C.X.; Mayer, E.L.; et al. Design of SERENA-6, a phase III switching trial of camizestrant in ESR1-mutant breast cancer during first-line treatment. Future Oncol. 2023, 19, 559–573. [Google Scholar] [CrossRef]
- Lawson, M.; Cureton, N.; Ros, S.; Cheraghchi-Bashi, A.; Urosevic, J.; D’Arcy, S.; Delpuech, O.; DuPont, M.; Fisher, D.I.; Gangl, E.T.; et al. The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance. Cancer Res. 2023, 83, 3989–4004. [Google Scholar] [CrossRef]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [Google Scholar] [CrossRef]
- Oliveira, M.; Pominchuk, D.; Nowecki, Z.; Hamilton, E.; Kulyaba, Y.; Andabekov, T.; Hotko, Y.; Melkadze, T.; Nemsadze, G.; Neven, P.; et al. Camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: Results of the randomized, multi-dose Phase 2 SERENA-2 trial. Lancet Oncol. 2024, 25, 1424–1439. [Google Scholar] [CrossRef]
- Gough, S.M.; Flanagan, J.J.; Teh, J.; Andreoli, M.; Rousseau, E.; Pannone, M.; Bookbinder, M.; Willard, R.; Davenport, K.; Bortolon, E.; et al. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Vahdat, L.; Han, H.S.; Ranciato, J.; Gedrich, R.; Keung, C.F.; Chirnomas, D.; Hurvitz, S. First-in-human safety and activity of ARV-471, a novel PROTAC® estrogen receptor degrader, in ER+/HER2− locally advanced or metastatic breast cancer. Cancer Res. 2022, 82 (Suppl. S4), PD13-08. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Schott, A.F.; Ma, C.; Hamilton, E.P.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.R.; Telli, M.L.; Mesias, J.A. ARV-471, a PROTAC estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: Phase 2 expansion (VERITAC) of a phase 1/2 study. In Proceedings of the 2022 San Antonio Breast Cancer Symposium, Los Angeles, CA, USA, 6–10 December 2022; p. GS3-03. [Google Scholar]
- Hamilton, E.P.; Ma, C.; De Laurentiis, M.; Iwata, H.; Hurvitz, S.A.; Wander, S.A.; Danso, M.; Lu, D.R.; Perkins Smith, J.; Liu, Y.; et al. VERITAC-2: A Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2− advanced breast cancer. Future Oncol. 2024, 20, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Parisian, A.D.; Barratt, S.A.; Hodges-Gallagher, L.; Ortega, F.E.; Pena, G.; Sapugay, J.; Robello, B.; Sun, R.; Kulp, D.; Palanisamy, G.S.; et al. Palazestrant (OP-1250), A Complete Estrogen Receptor Antagonist, Inhibits Wild-type and Mutant ER-positive Breast Cancer Models as Monotherapy and in Combination. Mol. Cancer Ther. 2024, 23, 285–300. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Endocrine Therapy Class and Study Details | Chemical Structure |
|---|---|---|
| AND019 | Selective ERα degrader (SERD) NCT05187832 Phase 1, Recruiting (Kind Pharmaceuticals) | NOT Disclosed |
| G1T48 | SERD NCT03455270 Phase 1, Completed (G1 Therapeutics) | 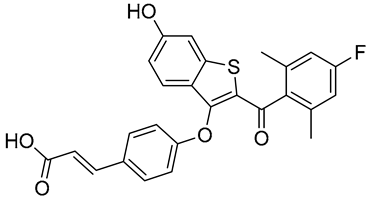 |
| D-0502 | SERD NCT03471663 Phase 1, Completed (InventisBio) |  |
| SIM0270 | SERD NCT05293964 Phase 1, Recruiting (Jiangsu Simcere Pharmaceutical) | NOT Disclosed |
| H3B-6545 | Selective ERα covalent antagonist (SERCA) NCT03250676 Phase 1, Completed (Eisai) | 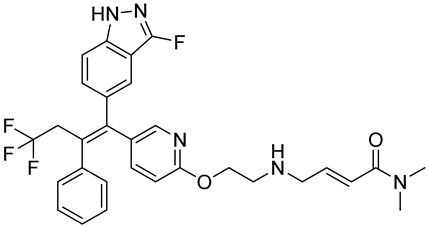 |
| ZN-c5 | SERD NCT03560531 Phase1/2, Completed (Zeno Alpha) |  |
| GDC-9545 | SERD NCT06065748 Phase 3, Recruiting (Hoffmann-La Roche) | 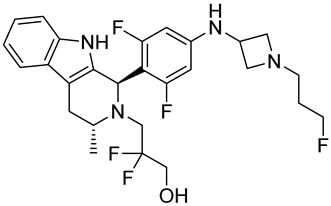 |
| LY3484356 | SERD NCT05514054 Phase 3, Recruiting (Eli Lilly and Company) |  |
| AZD9833 | SERD NCT04964934 Phase 3, Active, not recruiting (AstraZeneca) |  |
| OP-1250 | SERD NCT06016738 Phase 3, Recruiting (Olema Pharmaceuticals) | 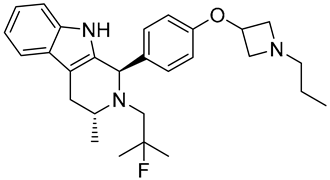 |
| ARV-471 (PF-07850327) | ER PROTAC degrader NCT05654623 Phase 3, Recruiting (Pfizer) | 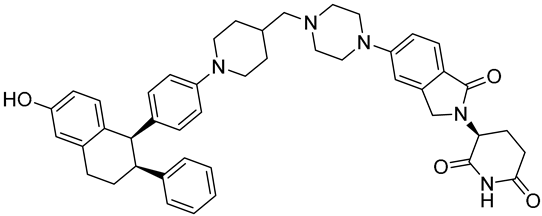 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palaniappan, M. Current Therapeutic Opportunities for Estrogen Receptor Mutant Breast Cancer. Biomedicines 2024, 12, 2700. https://doi.org/10.3390/biomedicines12122700
Palaniappan M. Current Therapeutic Opportunities for Estrogen Receptor Mutant Breast Cancer. Biomedicines. 2024; 12(12):2700. https://doi.org/10.3390/biomedicines12122700
Chicago/Turabian StylePalaniappan, Murugesan. 2024. "Current Therapeutic Opportunities for Estrogen Receptor Mutant Breast Cancer" Biomedicines 12, no. 12: 2700. https://doi.org/10.3390/biomedicines12122700
APA StylePalaniappan, M. (2024). Current Therapeutic Opportunities for Estrogen Receptor Mutant Breast Cancer. Biomedicines, 12(12), 2700. https://doi.org/10.3390/biomedicines12122700