
Dopamine Receptor Ligand Selectivity—An In Silico/In Vitro Insight

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ligand Selection for Combined In Silico/In Vitro Approach
2.3. Similarity Assessment—Tanimoto Scoring (TS) Matrix
2.4. Dataset Assembly for Molecular Docking (ChEMBL Validation)
2.5. Data Set Preparation for Molecular Docking
2.6. Molecular Docking Workflow
2.6.1. Molecular Docking—D1R
2.6.2. Molecular Dynamics Simulation (MDS)—D2R
2.6.3. Molecular Docking D2R
2.6.4. Molecular Docking D3R
2.7. DR Subtypes—BLASTP Alignment
2.8. Validation of the Molecular Docking Approach—ChEMBL Dataset(s)
2.9. Docking Analysis—Novel DR Ligands
2.10. HTRF-Based Receptor Binding Studies
2.11. Characterization of DR Carrier Cells (D1R and D3R)—Kd Determination
2.12. In Vitro Screening—Assessment of Compound Activity
2.13. Ligand Selection for Ki Determination
2.14. KI Determination for Selected Ligands
2.15. Data Processing, Representation and Analysis
3. Results
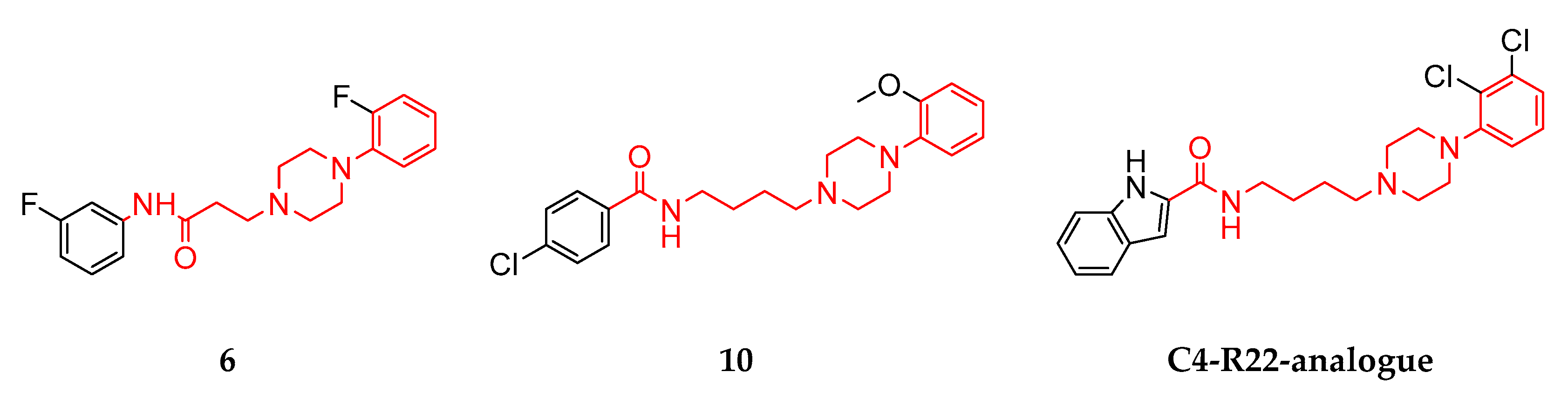
3.1. Structural Summary of the Investigated Ligands
3.2. In Vitro Compound Screening—An Assessment of DR Subtype Selectivity
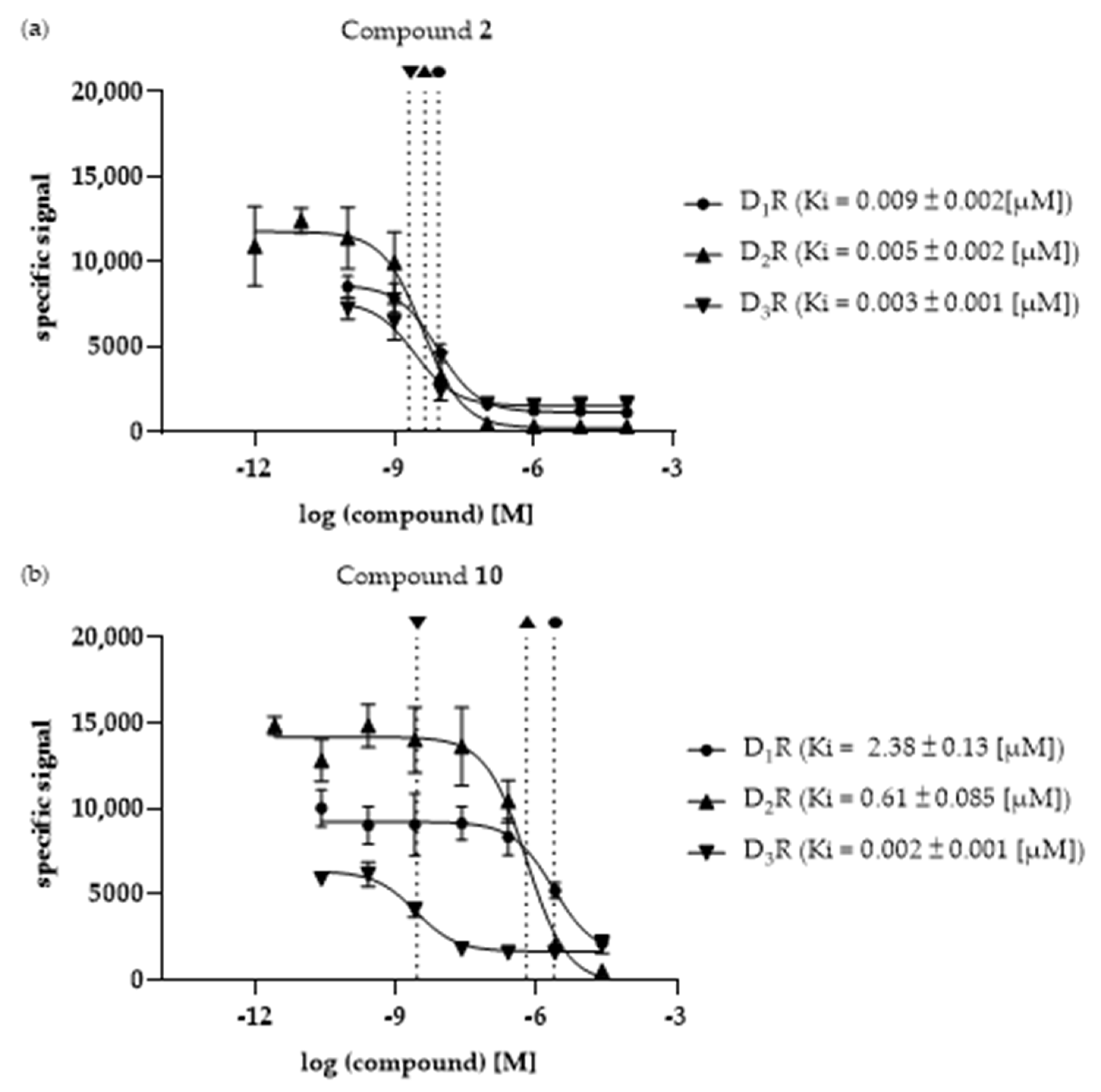
3.3. Ki Determination—Of the Selected Compounds at DR Subtypes
3.4. Dataset Assembly—In Silico Assessment
3.5. Validation of Molecular Docking
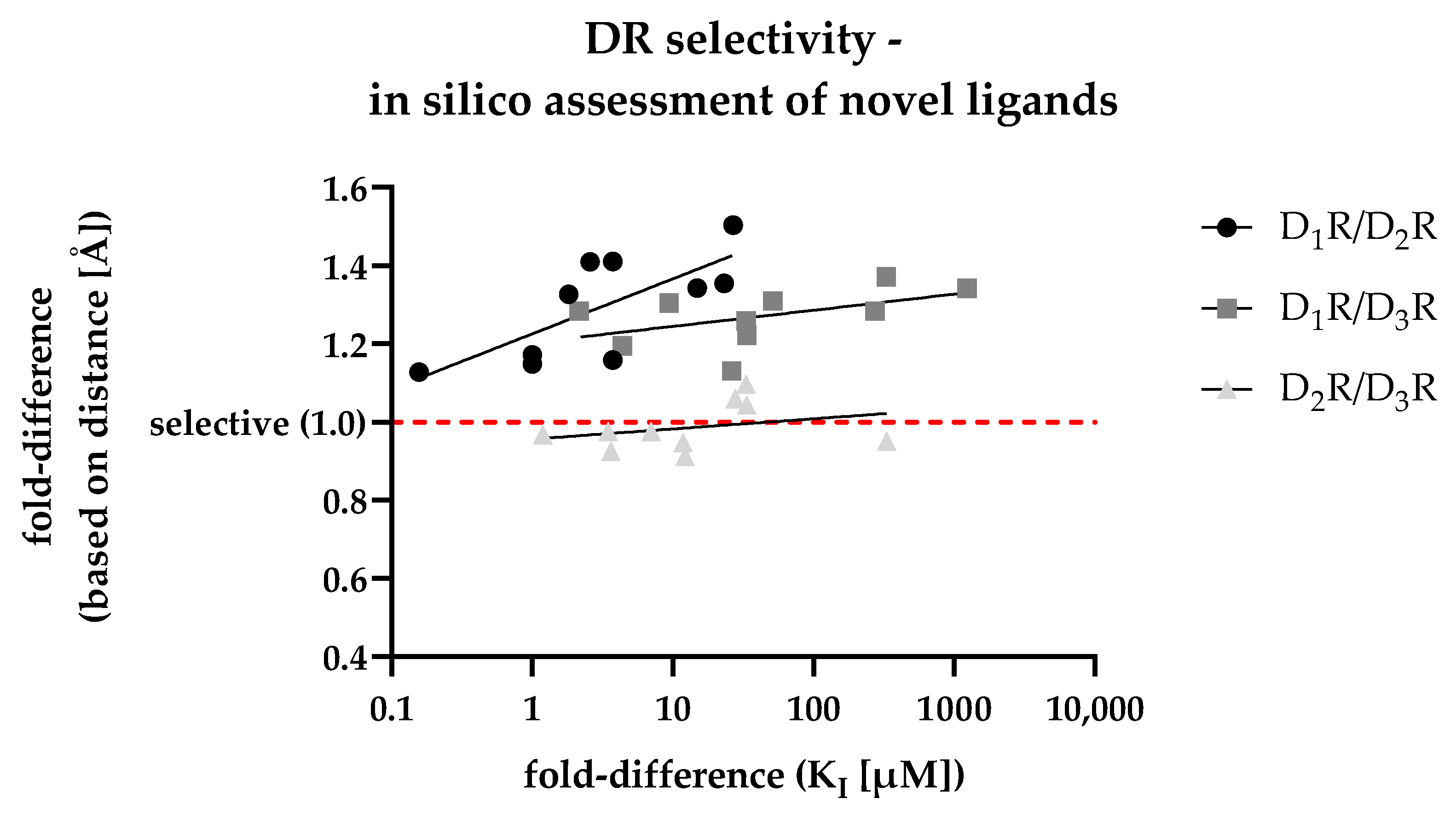
3.6. In Silico Assessment of DR Selectivity—Interaction with the SBP
3.7. Retrospective Analyis of the In Silico/In Vitro Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Stocchi, F.; Torti, M.; Fossati, C. Advances in dopamine receptor agonists for the treatment of Parkinson’s disease. Expert Opin. Pharmacother. 2016, 17, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.H.; Marques, T.R.; Jauhar, S.; Nour, M.M.; Goodwin, G.M.; Young, A.H.; Howes, O.D. The dopamine hypothesis of bipolar affective disorder: The state of the art and implications for treatment. Mol. Psychiatry 2017, 22, 666–679. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Kiss, B.; Laszlovsky, I.; Kramos, B.; Visegrady, A.; Levay, G.; Lendvai, B.; Roman, V. Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules 2021, 11, 104. [Google Scholar] [CrossRef]
- Prieto, G.A. Abnormalities of Dopamine D(3) Receptor Signaling in the Diseased Brain. J. Central Nerv. Syst. Dis. 2017, 9, 1–8. [Google Scholar] [CrossRef]
- Wang, Q.; Mach, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef]
- Murer, M.G.; Moratalla, R. Striatal Signaling in L-DOPA-Induced Dyskinesia: Common Mechanisms with Drug Abuse and Long Term Memory Involving D1 Dopamine Receptor Stimulation. Front. Neuroanat. 2011, 5, 51. [Google Scholar] [CrossRef]
- Berman, B.D. Neuroleptic malignant syndrome: A review for neurohospitalists. Neurohospitalist 2011, 1, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Moore, H.; Stott, L.; Holliday, N.; Javitch, J.A.; Lane, J.R.; Charlton, S.J. Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D(2) receptors. Nat. Commun. 2017, 8, 763. [Google Scholar] [CrossRef] [PubMed]
- Lao, C.L.; Kuo, Y.H.; Hsieh, Y.T.; Chen, J.C. Intranasal and subcutaneous administration of dopamine D3 receptor agonists functionally restores nigrostriatal dopamine in MPTP-treated mice. Neurotox. Res. 2013, 24, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Biswas, S.; Li, X.; Dutta, A.K.; Le, W. Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinson’s disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J. Neurosci. Res. 2010, 88, 2513–2523. [Google Scholar] [CrossRef]
- Antonini, A.; Barone, P.; Ceravolo, R.; Fabbrini, G.; Tinazzi, M.; Abbruzzese, G. Role of pramipexole in the management of Parkinson’s disease. CNS Drugs 2010, 24, 829–841. [Google Scholar] [CrossRef]
- Carnicella, S.; Drui, G.; Boulet, S.; Carcenac, C.; Favier, M.; Duran, T.; Savasta, M. Implication of dopamine D3 receptor activation in the reversion of Parkinson’s disease-related motivational deficits. Transl. Psychiatry 2014, 4, e401. [Google Scholar] [CrossRef]
- Millan, M.J.; Gobert, A.; Newman-Tancredi, A.; Lejeune, F.; Cussac, D.; Rivet, J.M.; Audinot, V.; Dubuffet, T.; Lavielle, G. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: I. Receptorial, electrophysiological and neurochemical profile compared with GR218,231 and L741,626. J. Pharmacol. Exp. Ther. 2000, 293, 1048–1062. [Google Scholar]
- Meltzer, H.Y. Cognitive factors in schizophrenia: Causes, impact, and treatment. CNS Spectr. 2004, 9, 15–24. [Google Scholar] [CrossRef]
- Jones-Tabah, J.; Mohammad, H.; Paulus, E.G.; Clarke, P.B.S.; Herbert, T.E. The Signaling and Pharmacology of the Dopamine D1 Receptor. Front. Cell. Neurosci. 2021, 15, 806618. [Google Scholar] [CrossRef]
- Felsing, D.E.; Jain, M.K.; Allen, J.A. Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr. Top. Med. Chem. 2019, 19, 1365–1380. [Google Scholar] [CrossRef]
- Girgis, R.R.; van Snellenberg, J.X.; Glass, A.; Kegeles, L.S.; Thompson, J.L.; Wall, M.; Cho, R.Y.; Carter, C.S.; Slifstein, M.; Abi-Dargham, A.; et al. A proof-of-concept, randomized controlled trial of DAR-0100A, a dopamine-1 receptor agonist, for cognitive enhancement in schizophrenia. J. Psychopharmacol. 2016, 30, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Rosell, D.R.; Zaluda, L.C.; McClure, M.M.; Strike, K.S.; Barch, D.M.; Harvey, P.D.; Girgis, R.R.; Hazlett, E.A.; Mailmann, R.B.; Abi-Dargham, A.; et al. Effects of the D1 dopamine receptor agonist dihydrexidine (DAR-0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology 2015, 40, 446–453. [Google Scholar] [CrossRef]
- Abi-Dargham, A.; Javitch, J.A.; Slifstein, M.; Anticevic, A.; Calkins, M.E.; Cho, Y.T.; Fonteneau, C.; Gil, R.; Girgis, R.; Gur, R.E.; et al. Dopamine D1R Receptor Stimulation as a Mechanistic Pro-cognitive Target for Schizophrenia. Schizophr. Bull. 2022, 48, 199–210. [Google Scholar] [CrossRef] [PubMed]
- O′Sullivan, G.J.; Dunleavy, M.; Hakansson, K.; Clementi, M.; Kinsella, A.; Croke, D.T.; Drago, J.; Fienberg, A.A.; Greengard, P.; Sibley, D.R.; et al. Dopamine D1 vs D5 receptor-dependent induction of seizures in relation to DARPP-32, ERK1/2 and GluR1-AMPA signalling. Neuropharmacology 2008, 54, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Girgis, R.R.; Gray, D.L.; Mailman, R.B. Novel Dopamine Therapeutics for Cognitive Deficits in Schizophrenia. Biol. Psychiatry 2017, 81, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.; Kiss, T.; Dlugolenski, K.; Johnson, D.E.; Gorczyca, R.R.; Kuszpit, K.; Harvey, B.D.; Stolyar, P.; Sukoff Rizzo, S.J.; Hoffmann, W.E.; et al. Characterization of PF-6142, a Novel, Non-Catecholamine Dopamine Receptor D1 Agonist, in Murine and Nonhuman Primate Models of Dopaminergic Activation. Front. Pharmacol. 2020, 11, 1005. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]
- Lian, P.; Xu, L.; Geng, C.; Qian, Y.; Li, W.; Zhen, X.; Fu, W. A computational perspective on drug discovery and signal transduction mechanism of dopamine and serotonin receptors in the treatment of schizophrenia. Curr. Pharm. Biotechnol. 2014, 15, 916–926. [Google Scholar] [CrossRef]
- Nikolic, K.; Mavridis, L.; Djikic, T.; Vucicevic, J.; Agbaba, D.; Yelekci, K.; Mitchell, J.B. Drug Design for CNS Diseases: Polypharmacological Profiling of Compounds Using Cheminformatic, 3D-QSAR and Virtual Screening Methodologies. Front. Neurosci. 2016, 10, 265. [Google Scholar] [CrossRef]
- Bueschbell, B.; Barreto, C.A.V.; Preto, A.J.; Schiedel, A.C.; Moreira, I.S. A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods. Molecules 2019, 24, 1196. [Google Scholar] [CrossRef] [PubMed]
- Floresca, C.Z.; Schetz, J.A. Dopamine receptor microdomains involved in molecular recognition and the regulation of drug affinity and function. J. Recept. Signal Transduct. Res. 2004, 24, 207–239. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Podlewska, S.; de Esch, I.J.P.; Bojarski, A.J.; Leurs, R.; Kooistra, A.J.; de Graaf, C. Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J. Med. Chem. 2019, 62, 3784–3839. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e18. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Beuming, T.; Banala, A.K.; Donthamsetti, P.; Pongetti, K.; LaBounty, A.; Levy, B.; Cao, J.; Michino, M.; Luedtke, R.R.; et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [Google Scholar] [CrossRef]
- Michino, M.; Donthamsetti, P.; Beuming, T.; Banala, A.; Duan, L.; Roux, T.; Han, Y.; Trinquet, E.; Newman, A.H.; Javitch, J.A.; et al. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol. 2013, 84, 854–864. [Google Scholar] [CrossRef]
- Robinson, S.W.; Jarvie, K.R.; Caron, M.G. High affinity agonist binding to the dopamine D3 receptor: Chimeric receptors delineate a role for intracellular domains. Mol. Pharmacol. 1994, 46, 352–356. [Google Scholar]
- Ishiki, H.M.; Filho, J.M.B.; da Silva, M.S.; Scotti, M.T.; Scotti, L. Computer-aided Drug Design Applied to Parkinson Targets. Curr. Neuropharmacol. 2018, 16, 865–880. [Google Scholar] [CrossRef]
- Elek, M.; Djokovic, N.; Frank, A.; Oljacic, S.; Zivkovic, A.; Nikolic, K.; Stark, H. Synthesis, in silico, and in vitro studies of novel dopamine D(2) and D(3) receptor ligands. Arch. Pharm. 2021, 354, e2000486. [Google Scholar] [CrossRef]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery—A review of theoretical aspects and recent applications. Curr. Chem. Genom. 2009, 3, 22–32. [Google Scholar] [CrossRef]
- Yasi, E.A.; Kruyer, N.S.; Peralta-Yahya, P. Advances in G protein-coupled receptor high-throughput screening. Curr. Opin. Biotechnol. 2020, 64, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Zell, L.; Lainer, C.; Kollar, J.; Temml, V.; Schuster, D. Identification of Novel Dopamine D(2) Receptor Ligands-A Combined In Silico/In Vitro Approach. Molecules 2022, 27, 4435. [Google Scholar] [CrossRef] [PubMed]
- Glen, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs 2006, 9, 199–204. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: Armonk, NY, USA, 1958. [Google Scholar]
- el Ahmad, Y.; Laurent, E.; Maillet, P.; Talab, A.; Teste, J.F.; Dokhan, R.; Tran, G.; Ollivier, R. New benzocycloalkylpiperazines, potent and selective 5-HT1A receptor ligands. J. Med. Chem. 1997, 40, 952–960. [Google Scholar] [CrossRef]
- Hubner, H.; Kraxner, J.; Gmeiner, P. Cyanoindole derivatives as highly selective dopamine D(4) receptor partial agonists: Solid-phase synthesis, binding assays, and functional experiments. J. Med. Chem. 2000, 43, 4563–4569. [Google Scholar] [CrossRef]
- Einsiedel, J.; Hubner, H.; Gmeiner, P. Benzamide bioisosteres incorporating dihydroheteroazole substructures: EPC synthesis and SAR leading to a selective dopamine D4 receptor partial agonist (FAUC 179). Bioorg. Med. Chem. Lett. 2001, 11, 2533–2536. [Google Scholar] [CrossRef]
- Lober, S.; Aboul-Fadl, T.; Hubner, H.; Gmeiner, P. Di- and trisubstituted pyrazolo[1,5-a]pyridine derivatives: Synthesis, dopamine receptor binding and ligand efficacy. Bioorg. Med. Chem. Lett. 2002, 12, 633–636. [Google Scholar] [CrossRef]
- Wittig, T.W.; Decker, M.; Lehmann, J. Dopamine/serotonin receptor ligands. 9. Oxygen-containing midsized heterocyclic ring systems and nonrigidized analogues. A step toward dopamine D5 receptor selectivity. J. Med. Chem. 2004, 47, 4155–4158. [Google Scholar] [CrossRef]
- Seong, C.M.; Park, W.K.; Park, C.M.; Kong, J.Y.; Park, N.S. Discovery of 3-aryl-3-methyl-1H-quinoline-2,4-diones as a new class of selective 5-HT6 receptor antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 738–743. [Google Scholar] [CrossRef]
- Enzensperger, C.; Muller, F.K.; Schmalwasser, B.; Wiecha, P.; Traber, H.; Lehmann, J. Dopamine/serotonin receptor ligands. 16.(1) Expanding dibenz[d,g]azecines to 11- and 12-membered homologues. Interaction with dopamine D(1)-D(5) receptors. J. Med. Chem. 2007, 50, 4528–4533. [Google Scholar] [CrossRef] [PubMed]
- Linz, S.; Muller, J.; Hubner, H.; Gmeiner, P.; Troschutz, R. Design, synthesis and dopamine D4 receptor binding activities of new N-heteroaromatic 5/6-ring Mannich bases. Bioorg. Med. Chem. 2009, 17, 4448–4458. [Google Scholar] [CrossRef] [PubMed]
- Banister, S.D.; Moussa, I.A.; Jorgensen, W.T.; Chua, S.W.; Kassiou, M. Molecular hybridization of 4-azahexacyclo[5.4.1.0(2,6).0(3,10).0(5,9).0(8,11)]dodecane-3-ol with sigma (sigma) receptor ligands modulates off-target activity and subtype selectivity. Bioorg. Med. Chem. Lett. 2011, 21, 3622–3626. [Google Scholar] [CrossRef] [PubMed]
- Robaa, D.; Enzensperger, C.; Eldin Abulazm, S.; Hefnawy, M.M.; El-Subbagh, H.I.; Wani, T.A.; Lehmann, J. Chiral indolo[3,2-f][3]benzazecine-type dopamine receptor antagonists: Synthesis and activity of racemic and enantiopure derivatives. J. Med. Chem. 2011, 54, 7422–7426. [Google Scholar] [CrossRef]
- Sampson, D.; Zhu, X.Y.; Eyunni, S.V.; Etukala, J.R.; Ofori, E.; Bricker, B.; Lamango, N.S.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Identification of a new selective dopamine D4 receptor ligand. Bioorg. Med. Chem. 2014, 22, 3105–3114. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, J.; Song, Z.; Guo, L.; Cai, W.; Wang, Y.; Zhen, X.; Zhang, A. Structural manipulation on the catecholic fragment of dopamine D(1) receptor agonist 1-phenyl-N-methyl-benzazepines. Eur. J. Med. Chem. 2014, 85, 16–26. [Google Scholar] [CrossRef]
- Madapa, S.; Harding, W.W. Semisynthetic Studies on and Biological Evaluation of N-Methyllaurotetanine Analogues as Ligands for 5-HT Receptors. J. Nat. Prod. 2015, 78, 722–729. [Google Scholar] [CrossRef]
- Lee, D.Y.W.; Liu, J.; Zhang, S.; Huang, P.; Liu-Chen, L.Y. Asymmetric total synthesis of tetrahydroprotoberberine derivatives and evaluation of their binding affinities at dopamine receptors. Bioorg. Med. Chem. Lett. 2017, 27, 1437–1440. [Google Scholar] [CrossRef]
- Martini, M.L.; Liu, J.; Ray, C.; Yu, X.; Huang, X.P.; Urs, A.; Urs, N.; McCorvy, J.D.; Caron, M.G.; Roth, B.L.; et al. Defining Structure-Functional Selectivity Relationships (SFSR) for a Class of Non-Catechol Dopamine D(1) Receptor Agonists. J. Med. Chem. 2019, 62, 3753–3772. [Google Scholar] [CrossRef]
- Heier, R.F.; Dolak, L.A.; Duncan, J.N.; Hyslop, D.K.; Lipton, M.F.; Martin, I.J.; Mauragis, M.A.; Piercey, M.F.; Nichols, N.F.; Schreur, P.J.; et al. Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo[4,5,1-ij]quinolin-5-amine and its metabolites. J. Med. Chem. 1997, 40, 639–646. [Google Scholar] [CrossRef]
- Thomas, C.; Hubner, H.; Gmeiner, P. Enantio- and diastereocontrolled dopamine D1, D2, D3 and D4 receptor binding of N-(3-pyrrolidinylmethyl)benzamides synthesized from aspartic acid. Bioorg. Med. Chem. Lett. 1999, 9, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, J.; Thomas, C.; Hubner, H.; Gmeiner, P. Phenyloxazoles and phenylthiazoles as benzamide bioisosteres: Synthesis and dopamine receptor binding profiles. Bioorg. Med. Chem. Lett. 2000, 10, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, T.; Hubner, H.; Gmeiner, P. Dopaminergic 7-aminotetrahydroindolizines: Ex-chiral pool synthesis and preferential D3 receptor binding. Bioorg. Med. Chem. Lett. 2001, 11, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, J.; Weber, K.; Thomas, C.; Lehmann, T.; Hubner, H.; Gmeiner, P. Stereocontrolled dopamine receptor binding and subtype selectivity of clebopride analogues synthesized from aspartic acid. Bioorg. Med. Chem. Lett. 2003, 13, 3293–3296. [Google Scholar] [CrossRef]
- Enguehard-Gueiffier, C.; Hubner, H.; el Hakmaoui, A.; Allouchi, H.; Gmeiner, P.; Argiolas, A.; Melis, M.R.; Gueiffier, A. 2-[(4-phenylpiperazin-1-yl)methyl]imidazo(di)azines as selective D4-ligands. Induction of penile erection by 2-[4-(2-methoxyphenyl)piperazin-1-ylmethyl]imidazo[1,2-a]pyridine (PIP3EA), a potent and selective D4 partial agonist. J. Med. Chem. 2006, 49, 3938–3947. [Google Scholar] [CrossRef]
- Tietze, R.; Lober, S.; Hubner, H.; Gmeiner, P.; Kuwert, T.; Prante, O. Discovery of a dopamine D4 selective PET ligand candidate taking advantage of a click chemistry based REM linker. Bioorg. Med. Chem. Lett. 2008, 18, 983–988. [Google Scholar] [CrossRef]
- Balle, T.; Perregaard, J.; Ramirez, M.T.; Larsen, A.K.; Soby, K.K.; Liljefors, T.; Andersen, K. Synthesis and structure-affinity relationship investigations of 5-heteroaryl-substituted analogues of the antipsychotic sertindole. A new class of highly selective alpha(1) adrenoceptor antagonists. J. Med. Chem. 2003, 46, 265–283. [Google Scholar] [CrossRef]
- Sromek, A.W.; Si, Y.G.; Zhang, T.; George, S.R.; Seeman, P.; Neumeyer, J.L. Synthesis and Evaluation of Fluorinated Aporphines: Potential Positron Emission Tomography Ligands for D2 Receptors. ACS Med. Chem. Lett. 2011, 2, 189–194. [Google Scholar] [CrossRef]
- Banerjee, A.; Maschauer, S.; Hubner, H.; Gmeiner, P.; Prante, O. Click chemistry based synthesis of dopamine D4 selective receptor ligands for the selection of potential PET tracers. Bioorg. Med. Chem. Lett. 2013, 23, 6079–6082. [Google Scholar] [CrossRef]
- Salama, I.; Lober, S.; Hubner, H.; Gmeiner, P. Synthesis and binding profile of haloperidol-based bivalent ligands targeting dopamine D(2)-like receptors. Bioorg. Med. Chem. Lett. 2014, 24, 3753–3756. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Hopkins, C.R. Return of D(4) Dopamine Receptor Antagonists in Drug Discovery. J. Med. Chem. 2017, 60, 7233–7243. [Google Scholar] [CrossRef] [PubMed]
- Glase, S.A.; Akunne, H.C.; Heffner, T.G.; Jaen, J.C.; MacKenzie, R.G.; Meltzer, L.T.; Pugsley, T.A.; Smith, S.J.; Wise, L.D. Aryl 1-but-3-ynyl-4-phenyl-1,2,3,6-tetrahydropyridines as potential antipsychotic agents: Synthesis and structure-activity relationships. J. Med. Chem. 1996, 39, 3179–3187. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, X.; Brodbeck, R.; Primus, R.; Braun, J.; Wasley, J.W.; Thurkauf, A. NGB 2904 and NGB 2849: Two highly selective dopamine D3 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2715–2718. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.M.; Bradley, P.A.; Gill, J.C.; Kerrigan, F.; Needham, P.L. N-Substituted (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine derivatives as D(2) antagonists/5-HT(1A) partial agonists with potential as atypical antipsychotic agents. J. Med. Chem. 1999, 42, 3342–3355. [Google Scholar] [CrossRef]
- Paul, N.M.; Taylor, M.; Kumar, R.; Deschamps, J.R.; Luedtke, R.R.; Newman, A.H. Structure-activity relationships for a novel series of dopamine D2-like receptor ligands based on N-substituted 3-aryl-8-azabicyclo[3.2.1]octan-3-ol. J. Med. Chem. 2008, 51, 6095–6109. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.S.; Majo, V.J.; Hsiung, S.C.; Millak, M.S.; Liu, K.P.; Tamir, H.; Prabhakaran, J.; Simpson, N.R.; van Heertum, R.L.; Mann, J.J.; et al. Synthesis and in vivo validation of [O-methyl-11C]2-4-[4-(7-methoxynaphthalen-1-yl)piperazin- 1-yl]butyl-4-methyl-2H-[1,2,4]triazine-3,5-dione: A novel 5-HT1A receptor agonist positron emission tomography ligand. J. Med. Chem. 2006, 49, 125–134. [Google Scholar] [CrossRef]
- Schlotter, K.; Boeckler, F.; Huber, H.; Gmeiner, P. Fancy bioisosteres: Novel paracyclophane derivatives as super-affinity dopamine D3 receptor antagonists. J. Med. Chem. 2006, 49, 3628–3635. [Google Scholar] [CrossRef]
- Newman, A.H.; Grundt, P.; Cyriac, G.; Deschamps, J.R.; Taylor, M.; Kumar, R.; Ho, D.; Luedtke, R.R. N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009, 52, 2559–2570. [Google Scholar] [CrossRef]
- Ortega, R.; Ravina, E.; Masaguer, C.F.; Areias, F.; Brea, J.; Loza, M.I.; Lopez, L.; Selent, J.; Pastor, M.; Sanz, F. Synthesis, binding affinity and SAR of new benzolactam derivatives as dopamine D3 receptor ligands. Bioorg. Med. Chem. Lett. 2009, 19, 1773–1778. [Google Scholar] [CrossRef]
- Skultety, M.; Hubner, H.; Lober, S.; Gmeiner, P. Bioisosteric replacement leading to biologically active [2.2]paracyclophanes with altered binding profiles for aminergic G-protein-coupled receptors. J. Med. Chem. 2010, 53, 7219–7228. [Google Scholar] [CrossRef]
- Hofling, S.B.; Maschauer, S.; Hubner, H.; Gmeiner, P.; Wester, H.J.; Prante, O.; Heinrich, M.R. Synthesis, biological evaluation and radiolabelling by 18F-fluoroarylation of a dopamine D3-selective ligand as prospective imaging probe for PET. Bioorg. Med. Chem. Lett. 2010, 20, 6933–6937. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Wu, Q.; Zhu, L.; Zheng, L.; Gao, B.; Zhen, X.; Zhang, A. Further SAR study on 11-O-substituted aporphine analogues: Identification of highly potent dopamine D3 receptor ligands. Bioorg. Med. Chem. 2011, 19, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Reinart-Okugbeni, R.; Ausmees, K.; Kriis, K.; Werner, F.; Rinken, A.; Kanger, T. Chemoenzymatic synthesis and evaluation of 3-azabicyclo[3.2.0]heptane derivatives as dopaminergic ligands. Eur. J. Med. Chem. 2012, 55, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective kappa opioid receptor agonist. J. Med. Chem. 2012, 55, 10302–10306. [Google Scholar] [CrossRef]
- Majo, V.J.; Milak, M.S.; Prabhakaran, J.; Mali, P.; Savenkova, L.; Simpson, N.R.; Mann, J.J.; Parsey, R.V.; Kumar, J.S. Synthesis and in vivo evaluation of [(18)F]2-(4-(4-(2-(2-fluoroethoxy)phenyl)piperazin-1-yl)butyl)-4-methyl-1,2,4-triazine-3,5(2H,4H)-dione ([(18)F]FECUMI-101) as an imaging probe for 5-HT1A receptor agonist in nonhuman primates. Bioorg. Med. Chem. 2013, 21, 5598–5604. [Google Scholar] [CrossRef]
- Abdelfattah, M.A.; Lehmann, J.; Abadi, A.H. Discovery of highly potent and selective D4 ligands by interactive SAR study. Bioorg. Med. Chem. Lett. 2013, 23, 5077–5081. [Google Scholar] [CrossRef]
- Insua, I.; Alvarado, M.; Masaguer, C.F.; Iglesias, A.; Brea, J.; Loza, M.I.; Carro, L. Synthesis and binding affinity of new 1,4-disubstituted triazoles as potential dopamine D(3) receptor ligands. Bioorg. Med. Chem. Lett. 2013, 23, 5586–5591. [Google Scholar] [CrossRef]
- van Wieringen, J.P.; Shalgunov, V.; Janssen, H.M.; Fransen, P.M.; Janssen, A.G.; Michel, M.C.; Booij, J.; Elsinga, P.H. Synthesis and characterization of a novel series of agonist compounds as potential radiopharmaceuticals for imaging dopamine D(2)/(3) receptors in their high-affinity state. J. Med. Chem. 2014, 57, 391–410. [Google Scholar] [CrossRef]
- Moller, D.; Kling, R.C.; Skultety, M.; Leuner, K.; Hubner, H.; Gmeiner, P. Functionally selective dopamine D(2), D(3) receptor partial agonists. J. Med. Chem. 2014, 57, 4861–4875. [Google Scholar] [CrossRef]
- Sampson, D.; Bricker, B.; Zhu, X.Y.; Peprah, K.; Lamango, N.S.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Further evaluation of the tropane analogs of haloperidol. Bioorg. Med. Chem. Lett. 2014, 24, 4294–4297. [Google Scholar] [CrossRef]
- Weichert, D.; Banerjee, A.; Hiller, C.; Kling, R.C.; Hubner, H.; Gmeiner, P. Molecular determinants of biased agonism at the dopamine D(2) receptor. J. Med. Chem. 2015, 58, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Jorg, M.; Kaczor, A.A.; Mak, F.S.; Lee, K.C.K.; Poso, A.; Miller, N.D.; Scammells, P.J.; Capuano, B. Investigation of novel ropinirole analogues: Synthesis, pharmacological evaluation and computational analysis of dopamine D-2 receptor functionalized congeners and homobivalent ligands. MedChemComm 2014, 5, 891–898. [Google Scholar] [CrossRef]
- Bartuschat, A.L.; Schellhorn, T.; Hubner, H.; Gmeiner, P.; Heinrich, M.R. Fluoro-substituted phenylazocarboxamides: Dopaminergic behavior and N-arylating properties for irreversible binding. Bioorg. Med. Chem. 2015, 23, 3938–3947. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.; Salama, I.; Kling, R.C.; Hubner, H.; Gmeiner, P. 1,4-Disubstituted aromatic piperazines with high 5-HT2A/D2 selectivity: Quantitative structure-selectivity investigations, docking, synthesis and biological evaluation. Bioorg. Med. Chem. 2015, 23, 6195–6209. [Google Scholar] [CrossRef] [PubMed]
- Weichert, D.; Stanek, M.; Hubner, H.; Gmeiner, P. Structure-guided development of dual beta2 adrenergic/dopamine D2 receptor agonists. Bioorg. Med. Chem. 2016, 24, 2641–2653. [Google Scholar] [CrossRef]
- Moller, D.; Banerjee, A.; Uzuneser, T.C.; Skultety, M.; Huth, T.; Plouffe, B.; Hubner, H.; Alzheimer, C.; Friedland, K.; Muller, C.P.; et al. Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1,5-a]pyridine Substructure. J. Med. Chem. 2017, 60, 2908–2929. [Google Scholar] [CrossRef]
- Mannel, B.; Dengler, D.; Shonberg, J.; Hubner, H.; Moller, D.; Gmeiner, P. Hydroxy-Substituted Heteroarylpiperazines: Novel Scaffolds for beta-Arrestin-Biased D(2)R Agonists. J. Med. Chem. 2017, 60, 4693–4713. [Google Scholar] [CrossRef]
- Mannel, B.; Hubner, H.; Moller, D.; Gmeiner, P. beta-Arrestin biased dopamine D2 receptor partial agonists: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2017, 25, 5613–5628. [Google Scholar] [CrossRef]
- Stossel, A.; Brox, R.; Purkayastha, N.; Hubner, H.; Hocke, C.; Prante, O.; Gmeiner, P. Development of molecular tools based on the dopamine D(3) receptor ligand FAUC 329 showing inhibiting effects on drug and food maintained behavior. Bioorg. Med. Chem. 2017, 25, 3491–3499. [Google Scholar] [CrossRef]
- Omran, A.; Eslamimehr, S.; Crider, A.M.; Neumann, W.L. Synthesis of 3-(3-hydroxyphenyl)pyrrolidine dopamine D(3) receptor ligands with extended functionality for probing the secondary binding pocket. Bioorg. Med. Chem. Lett. 2018, 28, 1897–1902. [Google Scholar] [CrossRef]
- Ashraf-Uz-Zaman, M.; Sajib, M.S.; Cucullo, L.; Mikelis, C.M.; German, N.A. Analogs of penfluridol as chemotherapeutic agents with reduced central nervous system activity. Bioorg. Med. Chem. Lett. 2018, 28, 3652–3657. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Taylor, M.; Griffin, S.A.; Amani, A.; Hayatshahi, H.; Korzekwa, K.; Ye, M.; Mach, R.H.; Liu, J.; Luedtke, R.R.; et al. Design, synthesis, and evaluation of N-(4-(4-phenyl piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamides as selective dopamine D(3) receptor ligands. Bioorg. Med. Chem. Lett. 2019, 29, 2690–2694. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhou, Q.; Yan, W.; Sun, J.; Kozikowski, A.P.; Zhao, S.; Huang, X.P.; Cheng, J. Design and Synthesis of Bitopic 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as Selective Dopamine D3 Receptor Ligands. J. Med. Chem. 2020, 63, 4579–4602. [Google Scholar] [CrossRef]
- Moritz, A.E.; Free, R.B.; Weiner, W.S.; Akano, E.O.; Gandhi, D.; Abramyan, A.; Keck, T.M.; Ferrer, M.; Hu, X.; Southall, N.; et al. Discovery, Optimization, and Characterization of ML417: A Novel and Highly Selective D(3) Dopamine Receptor Agonist. J. Med. Chem. 2020, 63, 5526–5567. [Google Scholar] [CrossRef] [PubMed]
- Battiti, F.O.; Newman, A.H.; Bonifazi, A. Exception That Proves the Rule: Investigation of Privileged Stereochemistry in Designing Dopamine D(3)R Bitopic Agonists. ACS Med. Chem. Lett. 2020, 11, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Bergauer, M.; Hubner, H.; Gmeiner, P. 2,4-Disubstituted pyrroles: Synthesis, traceless linking and pharmacological investigations leading to the dopamine D4 receptor partial agonist FAUC 356. Bioorg. Med. Chem. Lett. 2002, 12, 1937–1940. [Google Scholar] [CrossRef]
- Bettinetti, L.; Schlotter, K.; Hubner, H.; Gmeiner, P. Interactive SAR studies: Rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J. Med. Chem. 2002, 45, 4594–4597. [Google Scholar] [CrossRef]
- Hocke, C.; Prante, O.; Lober, S.; Hubner, H.; Gmeiner, P.; Kuwert, T. Synthesis and radioiodination of selective ligands for the dopamine D3 receptor subtype. Bioorg. Med. Chem. Lett. 2004, 14, 3963–3966. [Google Scholar] [CrossRef]
- Rodriguez Loaiza, P.; Lober, S.; Hubner, H.; Gmeiner, P. Click chemistry based solid phase supported synthesis of dopaminergic phenylacetylenes. Bioorg. Med. Chem. 2007, 15, 7248–7257. [Google Scholar] [CrossRef]
- Butini, S.; Gemma, S.; Campiani, G.; Franceschini, S.; Trotta, F.; Borriello, M.; Ceres, N.; Ros, S.; Coccone, S.S.; Bernetti, M.; et al. Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: Design, synthesis, and effects on behavior. J. Med. Chem. 2009, 52, 151–169. [Google Scholar] [CrossRef]
- Dorfler, M.; Tschammer, N.; Hamperl, K.; Hubner, H.; Gmeiner, P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 2008, 51, 6829–6838. [Google Scholar] [CrossRef] [PubMed]
- von Coburg, Y.; Kottke, T.; Weizel, L.; Ligneau, X.; Stark, H. Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett. 2009, 19, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Tschammer, N.; Elsner, J.; Goetz, A.; Ehrlich, K.; Schuster, S.; Ruberg, M.; Kuhhorn, J.; Thompson, D.; Whistler, J.; Hubner, H.; et al. Highly potent 5-aminotetrahydropyrazolopyridines: Enantioselective dopamine D3 receptor binding, functional selectivity, and analysis of receptor-ligand interactions. J. Med. Chem. 2011, 54, 2477–2491. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.B.; Bubser, M.; Jones, C.K.; Hayes, J.P.; Wepy, J.A.; Locuson, C.W.; Daniels, J.S.; Lindsley, C.W.; Hopkins, C.R. Discovery and Characterization of ML398, a Potent and Selective Antagonist of the D4 Receptor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Ponnala, S.; Kapadia, N.; Harding, W.W. Identification of tris-(phenylalkyl) amines as new selective h5-HT2B receptor antagonists. MedChemComm 2015, 6, 601–605. [Google Scholar] [CrossRef]
- Gadhiya, S.; Cordone, P.; Pal, R.K.; Gallicchio, E.; Wickstrom, L.; Kurtzmann, T.; Ramsey, S.; Harding, W.W. New Dopamine D3-Selective Receptor Ligands Containing a 6-Methoxy-1,2,3,4-tetrahydroisoquinolin-7-ol Motif. ACS Med. Chem. Lett. 2018, 9, 990–995. [Google Scholar] [CrossRef]
- Karki, A.; Namballa, H.K.; Alberts, I.; Harding, W.W. Structural manipulation of aporphines via C10 nitrogenation leads to the identification of new 5-HT(7A)R ligands. Bioorg. Med. Chem. 2020, 28, 115578. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Mao, C.; Krumm, B.E.; Zhou, X.E.; Tan, Y.; Huang, X.P.; Liu, Y.; Shen, D.D.; Jiang, Y.; et al. Structures of the human dopamine D3 receptor-G(i) complexes. Mol. Cell. 2021, 81, 1147–1159.e4. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. Dassault Systèmes, BIOVIA Discovery Studio, Release 2018; Dassault Systems: San Diego, CA, USA, 2018. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Joober, R.; Boksa, P. Clozapine: A distinct, poorly understood and under-used molecule. J. Psychiatry Neurosci. 2010, 35, 147–149. [Google Scholar] [CrossRef]
- Garcia-Romero, E.M.; López-López, E.; Soriano-Correa, C.; Medina-Franco, J.L.; Barrientos-Salcedo, C. Polypharmacological drug design opportunities against Parkinson’s disease. F1000Research 2022, 11, 1176. [Google Scholar] [CrossRef]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Leopoldo, M.; Tortorella, V. A structure-affinity relationship study on derivatives of N-[2-[4-(4-Chlorophenyl)piperazin-1-yl]ethyl]-3-methoxybenzamide, a high-affinity and selective D(4) receptor ligand. J. Med. Chem. 2000, 43, 270–277. [Google Scholar] [CrossRef]
- Smith, J.L.; Stein, D.A.; Shum, D.; Fischer, M.A.; Radu, C.; Bhinder, B.; Djaballah, H.; Nelson, J.A.; Fruh, K.; Hirsch, A.J. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J. Virol. 2014, 88, 5533–5542. [Google Scholar] [CrossRef]
- Campiani, G.; Nacci, V.; Bechelli, S.; Ciani, S.M.; Garofalo, A.; Fiorini, I.; Wikstrom, H.; de Boer, P.; Liao, Y.; Tepper, P.G.; et al. New antipsychotic agents with serotonin and dopamine antagonist properties based on a pyrrolo[2,1-b][1,3]benzothiazepine structure. J. Med. Chem. 1998, 41, 3763–3772. [Google Scholar] [CrossRef]
- Staron, J.; Kurczab, R.; Warszycki, D.; Satala, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT(6)/5-HT(2A) receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020, 185, 111857. [Google Scholar] [CrossRef]
- Peprah, K.; Zhu, X.Y.; Eyunni, S.V.; Setola, V.; Roth, B.L.; Ablordeppey, S.Y. Multi-receptor drug design: Haloperidol as a scaffold for the design and synthesis of atypical antipsychotic agents. Bioorg. Med. Chem. 2012, 20, 1291–1297. [Google Scholar] [CrossRef]
- Schoemaker, H.; Claustre, Y.; Fage, D.; Rouquier, L.; Chergui, K.; Curet, O.; Oblin, A.; Gonon, F.; Carter, C.; Benavides, J.; et al. Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J. Pharmacol. Exp. Ther. 1997, 280, 83–97. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Bragina, M.E.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Bowen, J.P.; Wyrick, S.D.; Hoffman, A.J.; Cory, M.; McPhail, A.T.; Mailman, R.B. Conformational analysis and molecular modeling of 1-phenyl-, 4-phenyl-, and 1-benzyl-1,2,3,4-tetrahydroisoquinolines as D1 dopamine receptor ligands. J. Med. Chem. 1989, 32, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, F.; Ponten, H.; Waters, N.; Waters, S.; Sonesson, C. Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J. Med. Chem. 2010, 53, 2510–2520. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, X.P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef]
- Nievergelt, A.; Huonker, P.; Schoop, R.; Altmann, K.H.; Gertsch, J. Identification of serotonin 5-HT1A receptor partial agonists in ginger. Bioorg. Med. Chem. 2010, 18, 3345–3351. [Google Scholar] [CrossRef]
- Maramai, S.; Gemma, S.; Brogi, S.; Campiani, G.; Butini, S.; Stark, H.; Brindisi, M. Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front. Neurosci. 2016, 10, 451. [Google Scholar] [CrossRef]
- Fan, L.; Tan, L.; Chen, Z.; Qi, J.; Nie, F.; Luo, Z.; Cheng, J.; Wang, S. Haloperidol bound D(2) dopamine receptor structure inspired the discovery of subtype selective ligands. Nat. Commun. 2020, 11, 1074. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Setting | Value |
|---|---|
| Flexible Sidechains | ASP103 R, 1 rotamer (free) |
| TRP285 R, 1 rotamer (free) | |
| PHE288 R, 1 rotamer (free) | |
| PHE289, 1 rotamer (free) | |
| ASN292 R, 1 rotamer (free) |
| Setting | Value |
|---|---|
| Flexible Sidechains | ASP110 R, 1 rotamer (free) |
| HIS349 R, 8 rotamers (constrained) |
| DR Subtype | |||
|---|---|---|---|
| D3R | D1R | D2R | Status |
| Val86 | Lys81 | Val91 | Conserved in D2like DRs |
| Leu89 | Ala84 | Leu94 | Conserved in D2like DRs |
| Gly94 | Gly88 | Gly98 | Conserved |
| Phe106 | Trp99 | Phe110 | Conserved in D2like DRs |
| Cys181 | Cys186 | Cys182 | Conserved |
| Cpd. | Normalized Decrease in Fluorescence (NDF) ± SD | ||
|---|---|---|---|
| D1R | D2R a | D3R | |
| Control | 1 | 1 | 1 |
| 1 | 3.47 ± 1.04 | 9.44 ± 5.97 | 3.82 ± 1.32 |
| 2 | 5.46 ± 1.97 | 40.41 ± 1.39 | 4.16 ± 1.08 |
| 3 | 1.78 ± 1.35 | 3.99 ± 2.58 | 3.96 ± 1.10 |
| 4 | 0.90 ± 0.31 | 0.90 ± 0.39 | 2.75 ± 0.61 |
| 5 | 1.26 ± 0.64 | 15.74 ± 18.15 | 2.65 ± 0.64 |
| 6 | 2.15 ± 1.16 | 8.18 ± 3.62 | 4.21 ± 0.79 |
| 7 | 1.57 ± 0.54 | 10.85 ± 4.93 | 3.79 ± 0.70 |
| 8 | 1.20 ± 0.59 | 1.10 ± 0.52 | 2.63 ± 0.52 |
| 9 | 1.71 ± 0.86 | 22.08 ± 6.62 | 3.97 ± 0.78 |
| 10 | 2.59 ± 1.04 | 22.89 ± 8.41 | 4.09 ± 1.07 |
| Cpd. | Ki [µM] | Selectivity | ||||
|---|---|---|---|---|---|---|
| D1R | D2R | D3R | D1R/ D2R | D1R/ D3R | D2R/ D3R | |
| 1 | 0.36 ± 0.009 | 2.36 ± 0.14 | 0.12 ± 0.048 | 0.15 | 3.06 | 19.8 |
| 2 | 0.009 ± 0.002 | 0.005 ± 0.002 a | 0.003 ± 0.001 | 1.95 | 3.23 | 1.66 |
| 3 | n.d. b | 4.66 ± 2.69 a | 0.38 ± 0.022 | >21.4 b | 262.2 | 12.2 |
| 4 | n.d. b | n.d. b | 3.68 ± 0.94 | - | >27.2 b | >27.2 b |
| 5 | 46.9 ± 27.4 | 10.95 ± 4.43 a | 2.25 ± 0.91 | 4.28 | 20.8 | 4.86 |
| 6 | 7.76 ± 4.41 | 1.35 ± 0.63 a | 0.37 ± 0.28 | 5.77 | 20.7 | 3.56 |
| 7 | 8.33 ± 2.17 | 2.78 ± 1.06 a | 0.68 ± 0.068 | 3.00 | 12.3 | 4.11 |
| 8 | n.d. b | n.d. b | 2.32 ± 0.92 | - | >43.1 b | >43.1 b |
| 9 | 9.46 ± 1.18 | 0.33 ± 0.093 a | 0.024 ± 0.003 | 28.6 | 395.1 | 13.8 |
| 10 | 2.38 ± 0.13 | 0.61 ± 0.085 | 0.002 ± 0.001 | 3.91 | 1031.4 | 263.7 |
| Dataset | Fold-Difference (Cons. Gly-COM)) | ||
|---|---|---|---|
| D1R/D2R | D1R/D3R | D2R/D3R | |
| D2R selective | 1.32 | 1.31 | 0.99 |
| D3R selective | 1.43 | 1.35 | 0.94 |
| D2like selective | 1.45 | 1.37 | 0.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zell, L.; Bretl, A.; Temml, V.; Schuster, D. Dopamine Receptor Ligand Selectivity—An In Silico/In Vitro Insight. Biomedicines 2023, 11, 1468. https://doi.org/10.3390/biomedicines11051468
Zell L, Bretl A, Temml V, Schuster D. Dopamine Receptor Ligand Selectivity—An In Silico/In Vitro Insight. Biomedicines. 2023; 11(5):1468. https://doi.org/10.3390/biomedicines11051468
Chicago/Turabian StyleZell, Lukas, Alina Bretl, Veronika Temml, and Daniela Schuster. 2023. "Dopamine Receptor Ligand Selectivity—An In Silico/In Vitro Insight" Biomedicines 11, no. 5: 1468. https://doi.org/10.3390/biomedicines11051468
APA StyleZell, L., Bretl, A., Temml, V., & Schuster, D. (2023). Dopamine Receptor Ligand Selectivity—An In Silico/In Vitro Insight. Biomedicines, 11(5), 1468. https://doi.org/10.3390/biomedicines11051468