
Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenic Properties of Amyloidogenic PPs
“Friends May Come and Go, but Enemies Accumulate” Thomas Jones
3. Physiological Roles of Amyloidogenic PPs
“You Never Really Know Your Friends from Your Enemies until the Ice Breaks.” Eskimo Proverb
4. Antimicrobial and Antiviral Properties of Amyloidogenic PPs
4.1. Antimicrobial Activity of Aβ Peptide and α-Synuclein
4.2. Antiviral Properties of Aβ and α-Synuclein
4.3. Antiviral Activity of α-Synuclein
5. Amyloidogenic ARS-COVID Proteins
6. Relationship between COVID-19 Viral Proteins and Amyloidogenic PPs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Wisniowski, B.; Wechalekar, A. Confirming the Diagnosis of Amyloidosis. Acta Haematol. 2020, 143, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G. Amyloids: From pathogenesis to function. Biochemistry 2015, 80, 1127–1144. [Google Scholar] [CrossRef]
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Weissman, J.S. Amyloid Structure: Conformational Diversity and Consequences. Annu. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid Fibril Formation by Aβ16-22, a Seven-Residue Fragment of the Alzheimer’s β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated with Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 3380. [Google Scholar] [CrossRef]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef]
- Muller-Spath, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Ruegger, S.; Reymond, L.; Nettels, D.; Schuler, B. From the Cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef]
- Surguchev, A.; Surguchov, A. Conformational Diseases: Looking into the eyes. Brain Res. Bull. 2010, 81, 12–24. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.; Balch, W.E.; Kelly, J.W. Functional Amyloid Formation within Mammalian Tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef]
- Guyonnet, B.; Egge, N.; Cornwall, G.A.; Ghule, P.N.; Xie, R.-L.; Medina, R.; Colby, J.L.; Jones, S.N.; Lian, J.B.; Stein, J.L.; et al. Functional Amyloids in the Mouse Sperm Acrosome. Mol. Cell. Biol. 2014, 34, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, M.; Meisl, G.; Taylor, J.D.; Michaels, T.C.T.; Levin, A.; Otzen, D.E.; Chapman, M.R.; Dobson, C.M.; Matthews, S.J.; Knowles, T.P.J. Physical Determinants of Amyloid Assembly in Biofilm Formation. mBio 2019, 10, e02279-18. [Google Scholar] [CrossRef]
- Hsu, J.T.-A.; Tien, C.-F.; Yu, G.-Y.; Shen, S.; Lee, Y.-H.; Hsu, P.-C.; Wang, Y.; Chao, P.-K.; Tsay, H.-J.; Shie, F.-S. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2021, 22, 8226. [Google Scholar] [CrossRef]
- Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell Responses to Extracellular α-Synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, F.; Lavatelli, F.; Di Silvestre, D.; Valentini, V.; Rossi, R.; Palladini, G.; Obici, L.; Verga, L.; Mauri, P.; Merlini, G. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood 2012, 119, 1844–1847. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C.; Sanchorawala, V. Amyloidomics comes of age. Blood 2012, 119, 1795–1796. [Google Scholar] [CrossRef]
- Bissig, C.; Rochin, L.; van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Hewitt, E.W. Why Are Functional Amyloids Non-Toxic in Humans? Biomolecules 2017, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the Transmembrane Domain Alter PMEL Amyloid Formation from Functional to Pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed]
- Iconomidou, V.A.; Chryssikos, G.D.; Gionis, V.; Galanis, A.S.; Cordopatis, P.; Hoenger, A.; Hamodrakas, S.J. Amyloid Fibril Formation Propensity is Inherent into the Hexapeptidetandemly Repeating Sequence of the Central Domain of Silk Moth Chorion Proteins of the A-family. J. Struct. Biol. 2006, 156, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Shumeyko, S.A.; Vikhlyantsev, I.M.; Bobylev, A.G. Amyloids: The History of Toxicity and Functionality. Biology 2021, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a Fragment of the Amyloid Precursor Associated with Alzheimer’s Disease. Science 1989, 245, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and Neurotoxic Effects of Amyloid β Protein: Reversal by Tachykinin Neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-beta and Tau Serve Antioxidant Functions in the Aging and Alzheimer. Brain. Free Radic. Biol. Med. 2002, 33, 1194–1199. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-associated Amyloid Beta-protein is an Antimicrobial Peptide. PLoS ONE 2010, 5, e950. [Google Scholar] [CrossRef]
- Paschou, M.; Liaropoulou, D.; Kalaitzaki, V.; Efthimiopoulos, S.; Papazafiri, P. Knockdown of Amyloid Precursor Protein Increases Ion Channel Expression and Alters Ca2+ Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 2302. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef]
- Rice, H.C.; De Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-β precursor protein functions as a GABA(B)R1a ligand to modulate synaptic transmission. Science 2019, 363, eaao4827. [Google Scholar] [CrossRef]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Kröhnert, K.; Schäfer, C.; Rammner, B.; Koo, S.J.; Claßen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N. Chapter 12—Functional Amyloids. In Biopolymer-Based Formulations; Pal, K., Banerjee, I., Sarkar, P., Kim, D., Deng, W.P., Dubey, N.K., Majumder, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 263–282. [Google Scholar]
- Alraawi, Z.; Banerjee, N.; Mohanty, S.; Kumar, T.K.S. Amyloidogenesis: What Do We Know So Far? Int. J. Mol. Sci. 2022, 23, 13970. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Chen, S.; Xu, C. Functions of amyloid precursor protein in metabolic diseases. Metabolism 2021, 115, 154454. [Google Scholar] [CrossRef]
- An, Y.A.; Crewe, C.; Asterholm, I.W.; Sun, K.; Chen, S.; Zhang, F.; Shao, M.; Funcke, J.-B.; Zhang, Z.; Straub, L.; et al. Dysregulation of amyloid precursor protein impairs adipose tissue mitochondrial function and promotes obesity. Nat. Metab. 2019, 1, 1243–1257. [Google Scholar] [CrossRef]
- Czeczor, J.K.; McGee, S.L. Emerging roles for the amyloid precursor protein and derived peptides in the regulation of cellular and systemic metabolism. J. Neuroendocr. 2017, 29, 12470. [Google Scholar] [CrossRef]
- Cai, W.; Li, L.; Sang, S.; Pan, X.; Zhong, C. Physiological Roles of β-amyloid in Regulating Synaptic Function: Implications for AD Pathophysiology. Neurosci. Bull. 2022, 1–20. [Google Scholar] [CrossRef]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic Activity Regulates Interstitial Fluid Amyloid-β Levels In Vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-β Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef]
- Garcia-Pardo, J.; Bartolomé-Nafría, A.; Chaves-Sanjuan, A.; Gil-Garcia, M.; Visentin, C.; Bolognesi, M.; Ricagno, S.; Ventura, S. Cryo-EM structure of hnRNPDL-2 fibrils, a functional amyloid associated with limb-girdle muscular dystrophy D3. Nat. Commun. 2023, 14, 239. [Google Scholar] [CrossRef] [PubMed]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Barbut, D.; Stolzenberg, E.; Zasloff, M. Gastrointestinal Immunity and Alpha-Synuclein. J. Park. Dis. 2019, 9, S313–S322. [Google Scholar] [CrossRef]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Vojtechova, I.; Machacek, T.; Kristofikova, Z.; Stuchlik, A.; Petrasek, T. Infectious origin of Alzheimer’s disease: Amyloid beta as a component of brain antimicrobial immunity. PLOS Pathog. 2022, 18, e1010929. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Eimer, W.A.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease: The potential therapeutic role of the natural antibiotic amyloid-β peptide. Neurodegener. Dis. Manag. 2016, 6, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3, Erratum in Neuron 2018, 100, 1527–1532.. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 2017, 31, 3216–3226. [Google Scholar] [CrossRef]
- Park, S.-C.; Moon, J.C.; Shin, S.Y.; Son, H.; Jung, Y.J.; Kim, N.-H.; Kim, Y.-M.; Jang, M.-K.; Lee, J.R. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem. Biophys. Res. Commun. 2016, 478, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional Alterations to the Nigrostriatal System in Mice Lacking All Three Members of the Synuclein Family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef]
- Papachroni, K.; Ninkina, N.; Wanless, J.; Kalofoutis, A.T.; Gnuchev, N.V.; Buchman, V.L. Peripheral Sensory Neurons Survive in the Absence of α- and γ-Synucleins. J. Mol. Neurosci. 2005, 25, 157–164. [Google Scholar] [CrossRef]
- Tomlinson, J.J.; Shutinoski, B.; Dong, L.; Meng, F.; Elleithy, D.; Lengacher, N.A.; Nguyen, A.P.; Cron, G.O.; Jiang, Q.; Roberson, E.D.; et al. Holocranohistochemistry enables the visualization of α-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. J. Neural Transm. 2017, 124, 721–738. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2016, 90, 2767–2782. [Google Scholar] [CrossRef]
- Bourgade, K.; Dupuis, G.; Frost, E.H.; Fülöp, T., Jr. Anti-Viral Properties of Amyloid-β Peptides. J. Alzheimer’s Dis. 2016, 54, 859–878. [Google Scholar] [CrossRef]
- Bourgade, K.; Garneau, H.; Giroux, G.; Le Page, A.Y.; Bocti, C.; Dupuis, G.; Frost, E.H.; Fülöp, T., Jr. β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 2015, 16, 85–98. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Kaushik, A.; Kujawska, M.; Ahmed, E.A.; Batiha, G.E. SARS-CoV-2 infection and Parkinson’s disease: Possible links and perspectives. J. Neurosci. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Lesteberg, K.; Beckham, J.D. Immunology of West Nile Virus Infection and the Role of Alpha-Synuclein as a Viral Restriction Factor. Viral Immunol. 2019, 32, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Labrie, V.; Brundin, P. Alpha-Synuclein to the Rescue: Immune Cell Recruitment by Alpha-Synuclein during Gastrointestinal Infection. J. Innate Immun. 2017, 9, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Austin, S.A.; Floden, A.M.; Murphy, E.J.; Combs, C.K. Alpha-synuclein expression modulates microglial activation phenotype. J. Neurosci. 2006, 26, 10558–10563. [Google Scholar] [CrossRef]
- Ait Wahmane, S.; Achbani, A.; Ouhaz, Z.; Elatiqi, M.; Belmouden, A.; Nejmeddine, M. The Possible Protective Role ofα-SynucleinAgainst Severe Acute Respiratory Syndrome Coronavirus 2 Infections in Patients With Parkinson’s Disease. Mov. Disord. 2020, 35, 1293–1294. [Google Scholar] [CrossRef]
- Mukherjee, S.K.; Knop, J.M.; Winter, R. Modulation of the conformational space of SARS- CoV-2 RNA Quadruplex RG-1 by cellular components and the amyloidogenic peptides α-synuclein and hIAPP. Chemistry 2022, 28, e202104182. [Google Scholar] [CrossRef]
- Monogue, B.; Chen, Y.; Sparks, H.; Behbehani, R.; Chai, A.; Rajic, A.J.; Massey, A.; Kleinschmidt-Demasters, B.K.; Vermeren, M.; Kunath, T.; et al. Alpha-synuclein supports type 1 interferon signalling in neurons and brain tissue. Brain 2022, 145, 3622–3636. [Google Scholar] [CrossRef]
- Seth, P.; Sarkar, N. A comprehensive mini-review on amyloidogenesis of different SARS-CoV-2 proteins and its effect on amyloid formation in various host proteins. 3 Biotech 2022, 12, 322. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.P. Hypothesis: AA amyloidosis is a factor causing systemic complications after coronavirus disease. Prion 2021, 15, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Tayeb-Fligelman, E.; Cheng, X.; Tai, C.; Bowler, J.T.; Griner, S.; Sawaya, M.R.; Seidler, P.M.; Jiang, Y.X.; Lu, J.; Rosenberg, G.M.; et al. Inhibition of amyloid formation of the Nucleoprotein of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Michiels, Y.; Houhou-Fidouh, N.; Collin, G.; Berger, J.; Kohli, E. Humoral Response Induced by Prime-Boost Vaccination with the ChAdOx1 nCoV-19 and mRNA BNT162b2 Vaccines in a Teriflunomide-Treated Multiple Sclerosis Patient. Vaccines 2021, 9, 1140. [Google Scholar] [CrossRef]
- Westermark, G.T.; Fändrich, M.; Westermark, P. AA Amyloidosis: Pathogenesis and Targeted Therapy. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional Amyloids As Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Charnley, M.; Islam, S.; Bindra, G.K.; Engwirda, J.; Ratcliffe, J.; Zhou, J.; Mezzenga, R.; Hulett, M.D.; Han, K.; Berryman, J.T.; et al. Neurotoxic amyloidogenic peptides in the proteome of SARS-COV2: Potential implications for neurological symptoms in COVID-19. Nat. Commun. 2022, 13, 3387. [Google Scholar] [CrossRef]
- Laudicella, R.; Burger, I.A.; Panasiti, F.; Longo, C.; Scalisi, S.; Minutoli, F.; Baldari, S.; Grimaldi, L.M.E.; Alongi, P. Subcutaneous Uptake on [18F]Florbetaben PET/CT: A Case Report of Possible Amyloid-Beta Immune-Reactivity After COVID-19 Vaccination. SN Compr. Clin. Med. 2021, 3, 2626–2628. [Google Scholar] [CrossRef] [PubMed]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2022, 13, 143–150. [Google Scholar] [CrossRef]
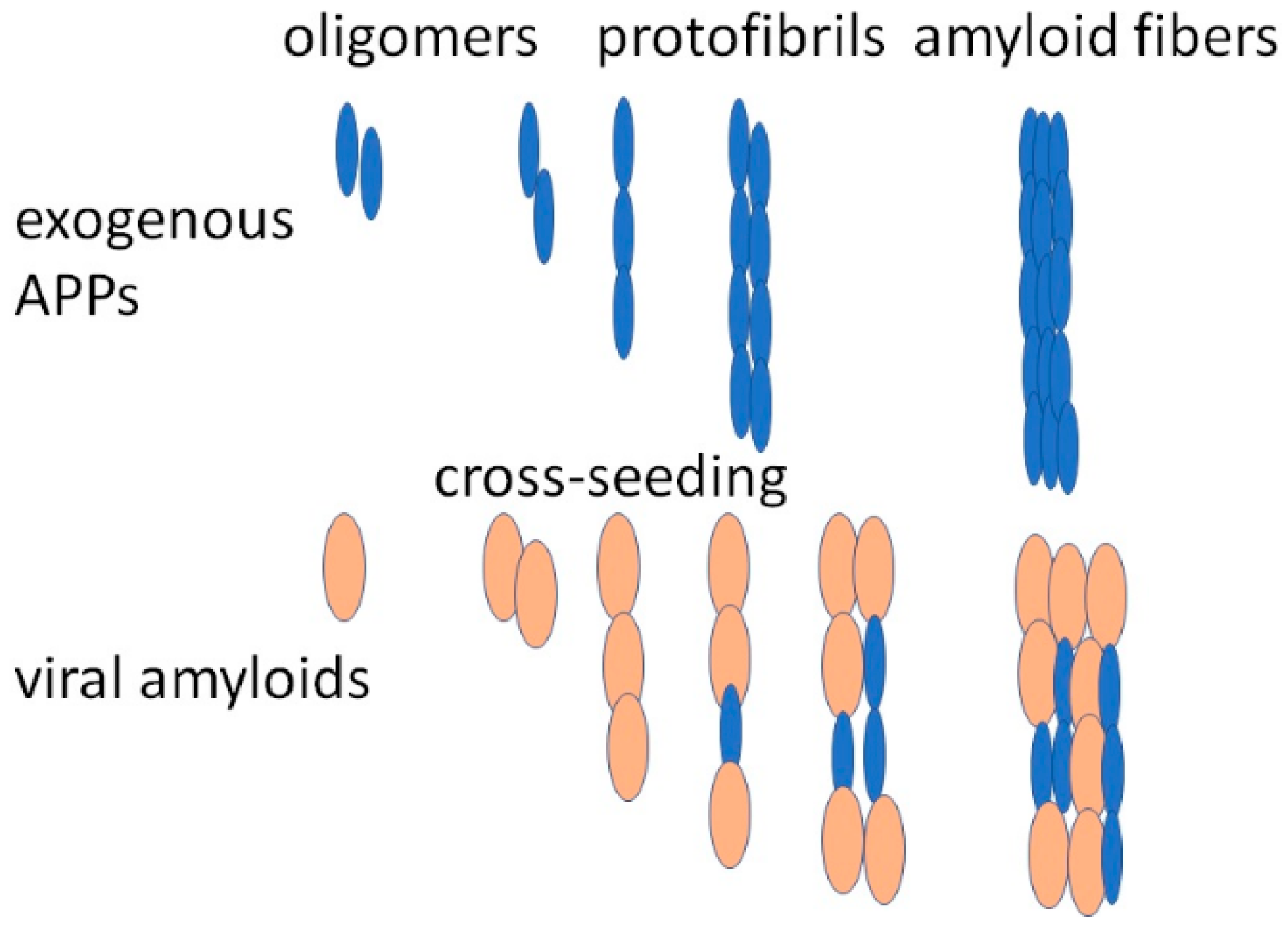
- Ge, W.-Y.; Deng, X.; Shi, W.-P.; Lin, W.-J.; Chen, L.-L.; Liang, H.; Wang, X.-T.; Zhang, T.-D.; Zhao, F.-Z.; Guo, W.-H.; et al. Amyloid Protein Cross-Seeding Provides a New Perspective on Multiple Diseases In Vivo. Biomacromolecules 2023, 24, 1–18. [Google Scholar] [CrossRef]
- Nyström, S.; Hammarström, P. Amyloidogenesis of SARS-CoV-2 Spike Protein. J. Am. Chem. Soc. 2022, 144, 8945–8950. [Google Scholar] [CrossRef]
- Rosen, B.; Kurtishi, A.; Vazquez-Jimenez, G.R.; Møller, S.G. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol. Neurobiol. 2021, 58, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Tavassoly, O.; Safavi, F.; Tavassoly, I. Seeding brain protein aggregation by SARS-CoV-2 as a possible long-term complication of COVID-19 infection. ACS Chem. Neurosci. 2020, 11, 3704–3706. [Google Scholar] [CrossRef]
- Mysiris, D.S.; Vavougios, G.D.; Karamichali, E.; Papoutsopoulou, S.; Stavrou, V.T.; Papayianni, E.; Boutlas, S.; Mavridis, T.; Foka, P.; Zarogiannis, S.G.; et al. Post-COVID-19 Parkinsonism and Parkinson’s Disease Pathogenesis: The Exosomal Cargo Hypothesis. Int. J. Mol. Sci. 2022, 23, 9739. [Google Scholar] [CrossRef]
- Bhardwaj, T.; Gadhave, K.; Kapuganti, S.K.; Kumar, P.; Brotzakis, Z.F.; Saumya, K.U.; Nayak, N.; Kumar, A.; Joshi, R.; Mukherjee, B.; et al. Amyloidogenic proteins in the SARS-CoV and SARS-CoV-2 proteomes. Nat Commun. 2023, 14, 945. [Google Scholar] [CrossRef] [PubMed]
- Olasunkanmi, O.I.; Chen, S.; Mageto, J.; Zhong, Z. Virus-Induced Cytoplasmic Aggregates and Inclusions Are Critical Cellular Regulatory and Antiviral Factors. Viruses 2020, 12, 399. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Are infections seeding some cases of Alzheimer’s disease? Nature 2020, 587, 22–25. [Google Scholar] [CrossRef]
- Surguchov, A. Pandemic meets epidemic: COVID-19 and neurodegenerative diseases. Biochem. Res. Internet 2020. Available online: https://www.hindawi.com/post/pandemic-meets-epidemic-covid-19-and-neurodegenerative-diseases/ (accessed on 16 April 2023).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surguchov, A.; Emamzadeh, F.N.; Titova, M.; Surguchev, A.A. Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era. Biomedicines 2023, 11, 1215. https://doi.org/10.3390/biomedicines11041215
Surguchov A, Emamzadeh FN, Titova M, Surguchev AA. Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era. Biomedicines. 2023; 11(4):1215. https://doi.org/10.3390/biomedicines11041215
Chicago/Turabian StyleSurguchov, Andrei, Fatemeh N. Emamzadeh, Mariya Titova, and Alexei A. Surguchev. 2023. "Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era" Biomedicines 11, no. 4: 1215. https://doi.org/10.3390/biomedicines11041215
APA StyleSurguchov, A., Emamzadeh, F. N., Titova, M., & Surguchev, A. A. (2023). Controversial Properties of Amyloidogenic Proteins and Peptides: New Data in the COVID Era. Biomedicines, 11(4), 1215. https://doi.org/10.3390/biomedicines11041215