

RAGE Inhibitors in Neurodegenerative Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Receptors for Advanced Glycation Endproducts (RAGE)
2.1. Soluble RAGE
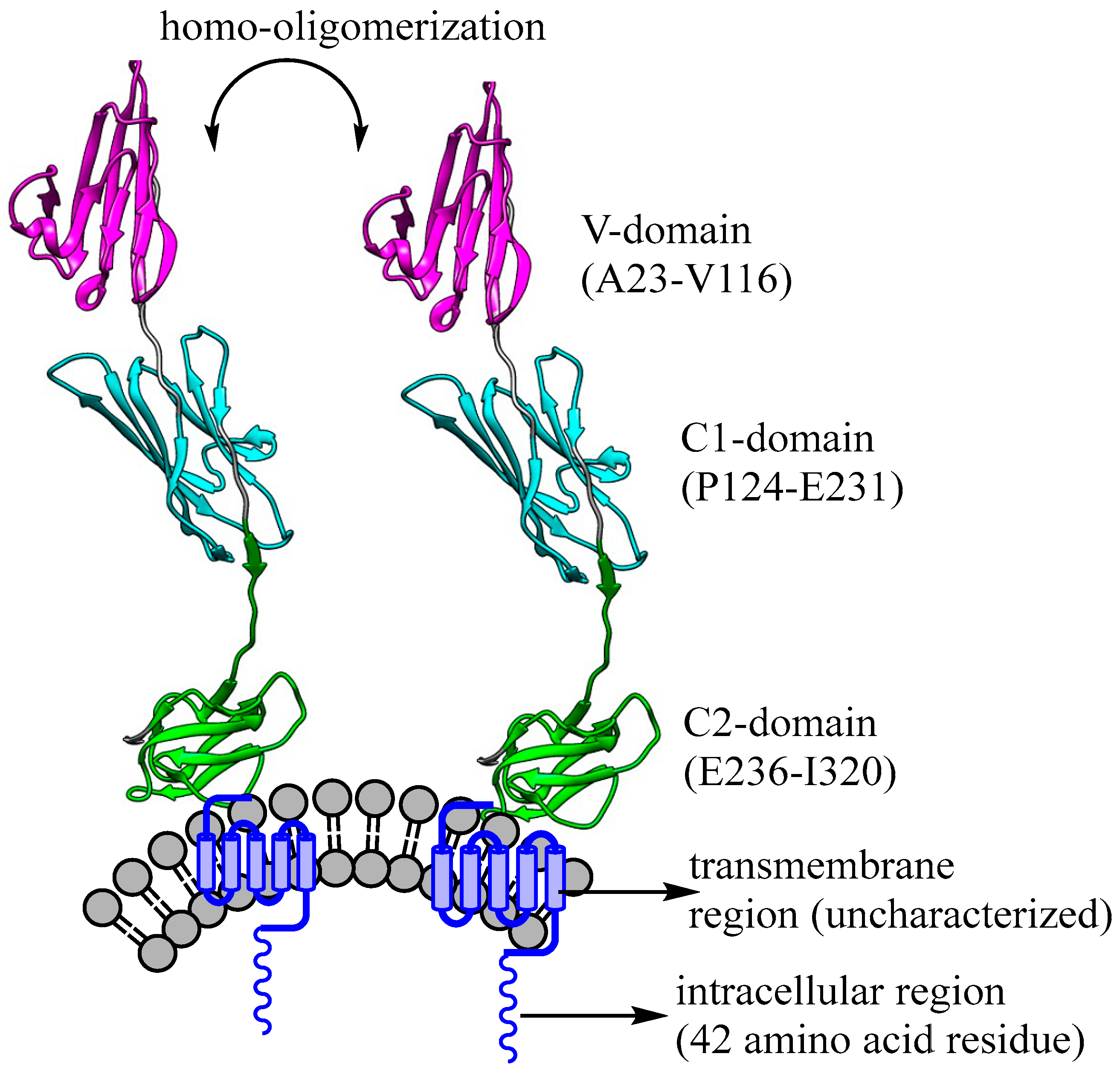
2.2. Structural Aspects of Receptors for Advanced Glycation Endproducts (RAGE)
3. RAGE and Gut Microbiota
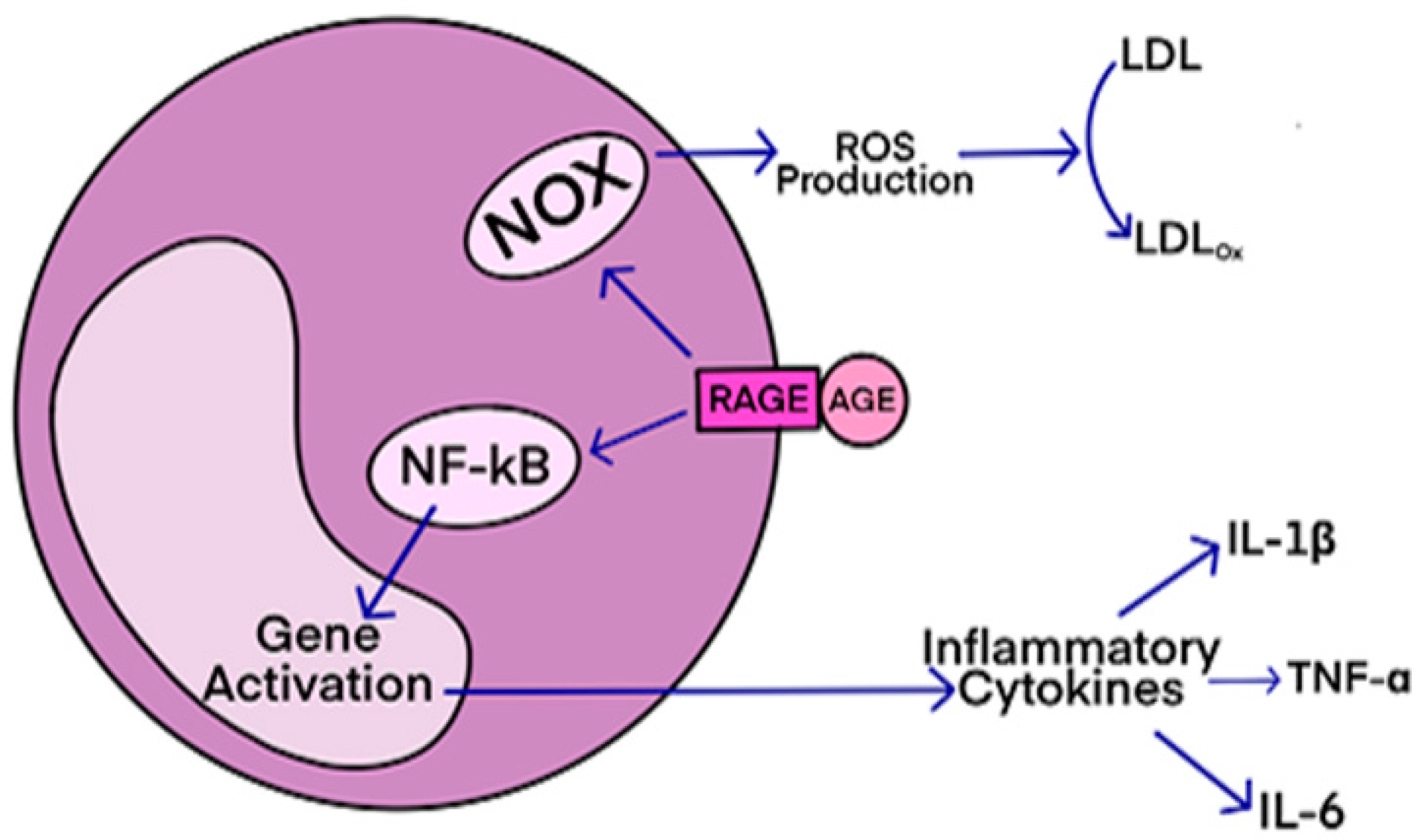
RAGE-Mediated Inflammatory Processes in Neurodegenerative Diseases
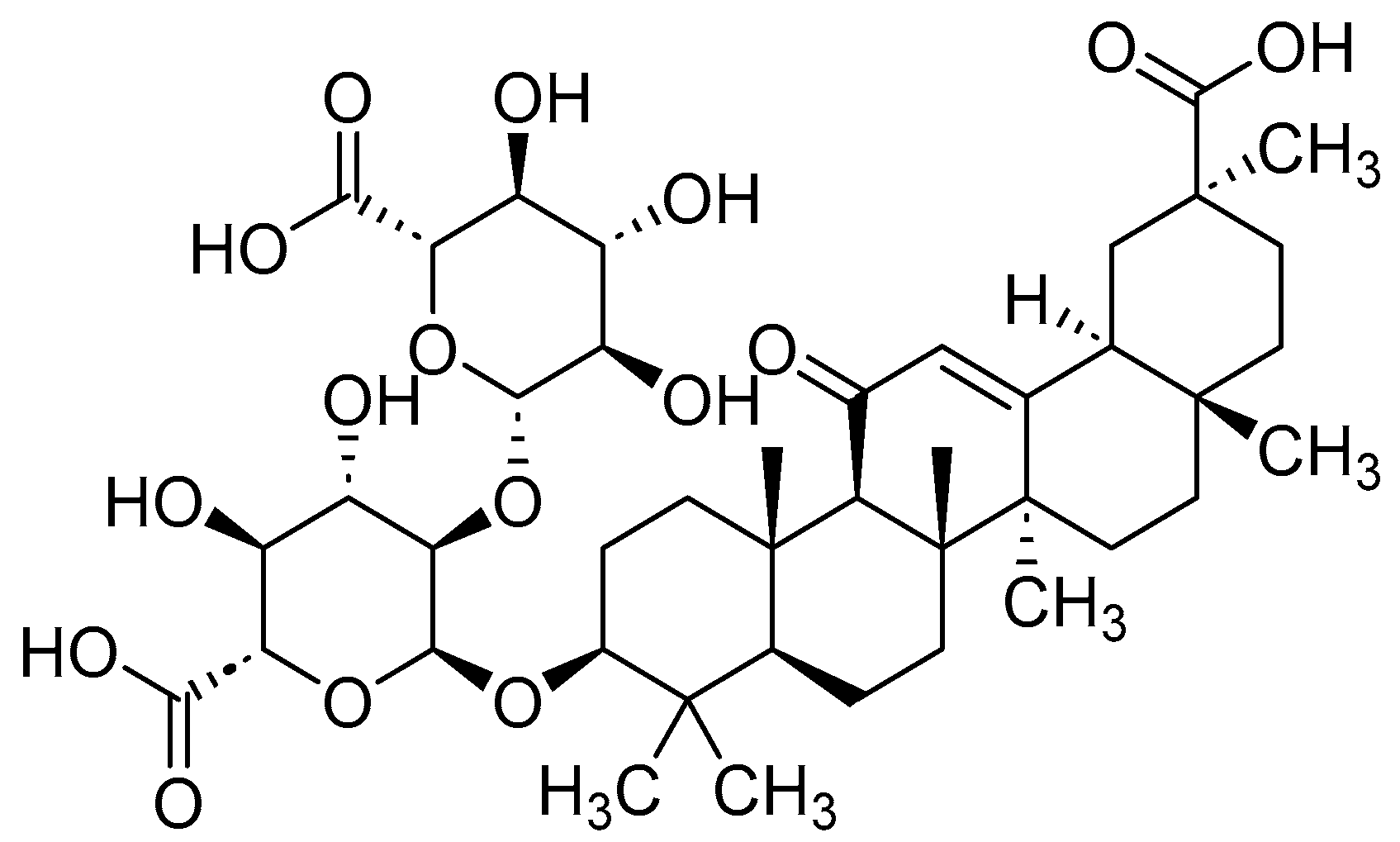
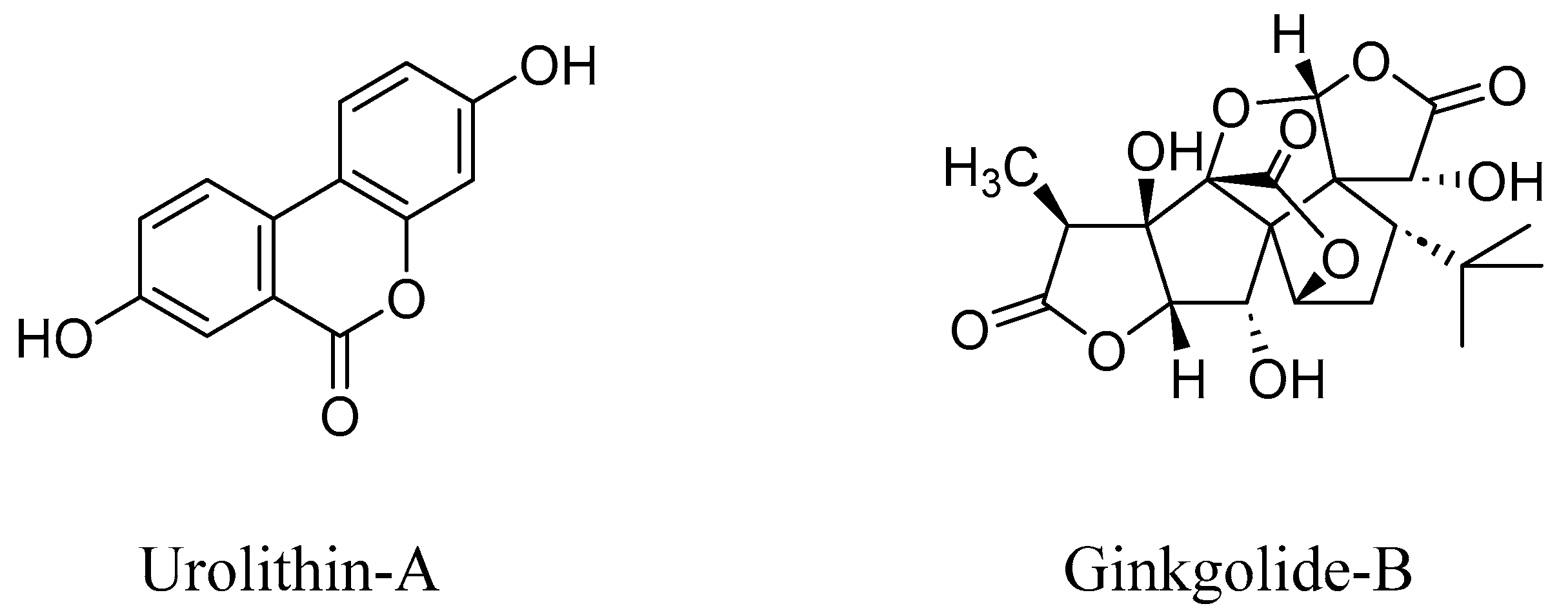
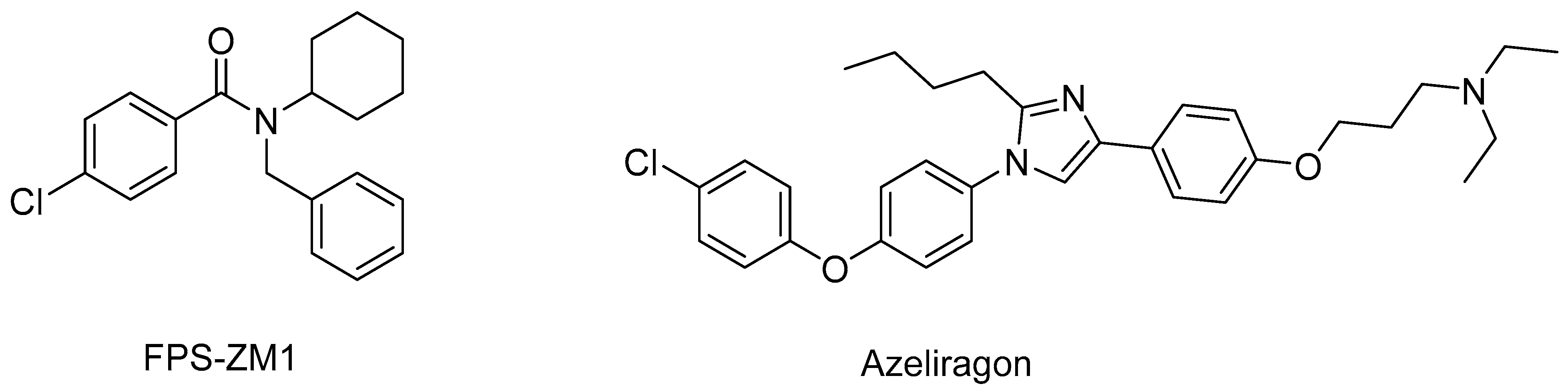
4. RAGE Antagonists
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reddy, V.P.; Aryal, P.; Darkwah, E.K. Advanced Glycation End Products in Health and Disease. Microorganisms 2022, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-A.; Wu, C.-H.; Yen, G.-C. Perspective of Advanced Glycation End Products on Human Health. J. Agric. Food Chem. 2018, 66, 2065–2070. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Li, L.; Li, Q.; Zhao, P.; Zhang, K.; Liu, C.; Cai, D.; Maegele, M.; Gu, Z.; Huang, Q. The role of S100B/RAGE-enhanced ADAM17 activation in endothelial glycocalyx shedding after traumatic brain injury. J. Neuroinflam. 2022, 19, 46. [Google Scholar] [CrossRef]
- Manivannan, S.; Marei, O.; Elalfy, O.; Zaben, M. Neurogenesis after traumatic brain injury—The complex role of HMGB1 and neuroinflammation. Neuropharmacology 2021, 183, 108400. [Google Scholar] [CrossRef]
- Saglam, E.; Zirh, S.; Aktas, C.C.; Muftuoglu, S.F.; Bilginer, B. Papaverine provides neuroprotection by suppressing neuroinflammation and apoptosis in the traumatic brain injury via RAGE-NF-<kappa>B pathway. J. Neuroimmunol. 2021, 352, 577476. [Google Scholar]
- Nowicka, N.; Szymanska, K.; Juranek, J.; Zglejc-Waszak, K.; Korytko, A.; Zalecki, M.; Chmielewska-Krzesinska, M.; Wasowicz, K.; Wojtkiewicz, J. The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. Int. J. Mol. Sci. 2022, 23, 2184. [Google Scholar] [CrossRef]
- Faruqui, T.; Khan, M.S.; Akhter, Y.; Khan, S.; Rafi, Z.; Saeed, M.; Han, I.; Choi, E.-H.; Yadav, D.K. RAGE Inhibitors for Targeted Therapy of Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 266. [Google Scholar] [CrossRef]
- Adeshara, K.A.; Bangar, N.; Diwan, A.G.; Tupe, R.S. Plasma glycation adducts and various RAGE isoforms are intricately associated with oxidative stress and inflammatory markers in type 2 diabetes patients with vascular complications. Diabetes Metab. Syndr. 2022, 16, 102441. [Google Scholar] [CrossRef]
- Vulichi, S.R.; Runthala, A.; Begari, N.; Rupak, K.; Chunduri, V.R.; Kapur, S.; Chippada, A.R.; Sistla, D.S.M. Type-2 diabetes mellitus-associated cancer risk: In pursuit of understanding the possible link. Diabetes Metab. Syndr. 2022, 16, 102591. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Hudson, B.I.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef]
- Patil, G.; Kulsange, S.; Kazi, R.; Chirmade, T.; Kale, V.; Mote, C.; Aswar, M.; Koratkar, S.; Agawane, S.; Kulkarni, M. Behavioral and Proteomic Studies Reveal Methylglyoxal Activate Pathways Associated with Alzheimer’s Disease. ACS Pharmacol. Transl. Sci. 2023, 6, 65–75. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Biswas, S. AGE/Non-AGE Glycation: An Important Event in Rheumatoid Arthritis Pathophysiology. Inflammation 2022, 45, 477–496. [Google Scholar] [CrossRef]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef]
- Jessop, F.; Schwarz, B.; Scott, D.; Roberts, L.M.; Bohrnsen, E.; Hoidal, J.R.; Bosio, C.M. Impairing RAGE signaling promotes survival and limits disease pathogenesis following SARS-CoV-2 infection in mice. JCI Insight 2022, 7, e155896. [Google Scholar] [CrossRef]
- Kozlyuk, N.; Gilston, B.A.; Salay, L.E.; Gogliotti, R.D.; Christov, P.P.; Kim, K.; Ovee, M.; Waterson, A.G.; Chazin, W.J. A fragment-based approach to discovery of Receptor for Advanced Glycation End products inhibitors. Proteins Struct. Funct. Bioinf. 2021, 89, 1399–1412. [Google Scholar] [CrossRef]
- Singh, H.; Agrawal, D.K. Therapeutic potential of targeting the receptor for advanced glycation end products (RAGE) by small molecule inhibitors. Drug Dev. Res. 2022, 83, 1257–1269. [Google Scholar] [CrossRef]
- Ma, S.; Nakamura, Y.; Hisaoka-Nakashima, K.; Morioka, N. Blockade of receptor for advanced glycation end-products with azeliragon ameliorates streptozotocin-induced diabetic neuropathy. Neurochem. Int. 2023, 163, 105470. [Google Scholar] [CrossRef]
- Singh, H.; Agrawal, D.K. Therapeutic Potential of Targeting the HMGB1/RAGE Axis in Inflammatory Diseases. Molecules 2022, 27, 7311. [Google Scholar] [CrossRef]
- Curran, C.S.; Kopp, J.B. RAGE pathway activation and function in chronic kidney disease and COVID-19. Front. Med. 2022, 9, 970423. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, Y.; Huang, Y.; Deng, H. Pathophysiology of RAGE in inflammatory diseases. Front. Immunol. 2022, 13, 931473. [Google Scholar] [CrossRef] [PubMed]
- Twarda-Clapa, A.; Olczak, A.; Bialkowska, A.M.; Koziolkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Obrenovich, M.E.; Atwood, C.S.; Perry, G.; Smith, M.A. Involvement of Maillard reactions in Alzheimer disease. Neurotoxic. Res. 2002, 4, 191–209. [Google Scholar] [CrossRef]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discovery Today 2006, 11, 646–654. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e912. [Google Scholar] [CrossRef]
- Inan-Eroglu, E.; Ayaz, A.; Buyuktuncer, Z. Formation of advanced glycation endproducts in foods during cooking process and underlying mechanisms: A comprehensive review of experimental studies. Nutr. Res. Rev. 2020, 33, 77–89. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, S.; Shi, Y.; Wang, P.; Wu, Y.; Li, G. Dietary advanced glycation end products (dAGEs): An insight between modern diet and health. Food Chem. 2023, 415, 135735. [Google Scholar] [CrossRef]
- Li, Y.; Peng, Y.; Shen, Y.; Zhang, Y.; Liu, L.; Yang, X. Dietary polyphenols: Regulate the advanced glycation end products-RAGE axis and the microbiota-gut-brain axis to prevent neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2022, 1–27. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Galasko, D.; Bell, J.; Mancuso, J.Y.; Kupiec, J.W.; Sabbagh, M.N.; van Dyck, C.; Thomas, R.G.; Aisen, P.S. Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 2014, 82, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Z.; Kouznetsova, V.L.; Tsigelny, I.F. Deep-learning- and pharmacophore-based prediction of RAGE inhibitors. Phys. Biol. 2020, 17, 036003. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Lue, L.F.; Paul, G.; Patel, A.; Sabbagh, M.N. Receptor for advanced glycation endproduct modulators: A new therapeutic target in Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Zhang, S.; Zeng, S.; Tong, Y.; Liu, J.; Liu, C.; Li, D. Interaction of RAGE with α-synuclein fibrils mediates inflammatory response of microglia. Cell Rep. 2022, 40, 111401. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Semple, B.; Piperi, C.; Othman, I.; Shaikh, M.F. Potential Neuroprotective Effect of the HMGB1 Inhibitor Glycyrrhizin in Neurological Disorders. ACS Chem. Neurosci. 2020, 11, 485–500. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Implication of HMGB1 signaling pathways in Amyotrophic lateral sclerosis (ALS): From molecular mechanisms to pre-clinical results. Pharmacol. Res. 2020, 156, 104792. [Google Scholar] [CrossRef]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.E.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998. [Google Scholar] [CrossRef]
- Bonnardel-Phu, E.; Wautier, J.-L.; Schmidt, A.M.; Avila, C.; Vicaut, E. Acute modulation of albumin microvascular leakage by advanced glycation end products in microcirculation of diabetic rats in vivo. Diabetes 1999, 48, 2052–2058. [Google Scholar] [CrossRef]
- Cooke, C.-L.M.; Brockelsby, J.C.; Baker, P.N.; Davidge, S.T. The receptor for advanced glycation end products (RAGE) is elevated in women with preeclampsia. Hypertens. Pregnancy 2003, 22, 173–184. [Google Scholar] [CrossRef]
- Khaket, T.P.; Kang, S.C.; Mukherjee, T.K. The Potential of Receptor for Advanced Glycation End Products (RAGE) as a Therapeutic Target for Lung Associated Diseases. Curr. Drug Targets 2019, 20, 679–689. [Google Scholar] [CrossRef]
- Nicholas, A.; Pei, T.; Bush, E.W.; Kasahara, D.I.; Schienebeck, C.M. Double Stranded RNAi Agents for Inhibiting Expression of RAGE or Receptor for Advanced Glycation End-Products and Compositions to Treat Respiratory or Ocular Disease, COPD, Pulmonary Inflammation, Severe Asthma, etc. WO2022216920, 13 October 2022. [Google Scholar]
- Schmidt, A.M.; Vianna, M.; Gerlach, M.; Brett, J.; Ryan, J.; Kao, J.; Esposito, C.; Hegarty, H.; Hurley, W. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 1992, 267, 14987. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.D.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Li, F.; Carey, L.B.; Sun, C.; Ling, K.; Tachikawa, H.; Fujita, M.; Gao, X.-D.; Nakanishi, H. Receptor for advanced glycation end-products (RAGE) mediates phagocytosis in nonprofessional phagocytes. Commun. Biol. 2022, 5, 824. [Google Scholar] [CrossRef]
- Wautier, J.-L.; Wautier, M.-P. Cellular and molecular aspects of blood cell-endothelium interactions in vascular disorders. Int. J. Mol. Sci. 2020, 21, 5315. [Google Scholar] [CrossRef]
- Wuren, T.; Huecksteadt, T.; Beck, E.; Warren, K.; Hoidal, J.; Ostrand-Rosenberg, S.; Sanders, K. The receptor for advanced glycation endproducts (RAGE) decreases survival of tumor-bearing mice by enhancing the generation of lung metastasis-associated myeloid-derived suppressor cells. Cell. Immunol. 2021, 365, 104379. [Google Scholar] [CrossRef]
- Lin, F.; Shan, W.; Zheng, Y.; Pan, L.; Zuo, Z. Toll-like receptor 2 activation and up-regulation by high mobility group box-1 contribute to post-operative neuroinflammation and cognitive dysfunction in mice. J. Neurochem. 2021, 158, 328–341. [Google Scholar] [CrossRef]
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol. Ther. 2014, 141, 347–357. [Google Scholar] [CrossRef]
- Jangde, N.; Ray, R.; Rai, V. RAGE and its ligands: From pathogenesis to therapeutics. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 555–575. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Piperi, C.; Gargalionis, A.N.; Dalagiorgou, G.; Spilioti, E.; Korkolopoulou, P.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-κB and JNK-AP-1 signaling pathways. Cell. Mol. Life Sci. 2016, 73, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Qiao, P.-F.; Wan, C.-Q.; Cai, M.; Zhou, N.-K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Chang, J.; Liu, J.; Liu, Y.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble RAGE attenuates myocardial I/R injuries via FoxO3-Bnip3 pathway. Cell. Mol. Life Sci. 2022, 79, 269. [Google Scholar] [CrossRef] [PubMed]
- Metz, V.V.; Kojro, E.; Rat, D.; Postina, R. Induction of RAGE shedding by activation of G protein-coupled receptors. PLoS ONE 2012, 7, e41823. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Deng, C.-Q.; Wang, J.; Deng, X.-J.; Xiao, Q.; Li, Y.; He, Q.; Fan, W.-H.; Quan, F.-Y.; Zhu, Y.-P.; et al. Plasma levels of soluble receptor for advanced glycation end products in Alzheimer’s disease. Int. J. Neurosci. 2017, 127, 454–458. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.M.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Swanner, J.; Shim, J.S.; Rivera-Caraballo, K.A.; Vazquez-Arreguin, K.; Hong, B.; Bueso-Perez, A.J.; Lee, T.J.; Banasavadi-Siddegowda, Y.K.; Kaur, B.; Yoo, J.Y. esRAGE-expressing oHSV enhances anti-tumor efficacy by inhibition of endothelial cell activation. Mol. Ther. Oncolytics 2023, 28, 171–181. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Zeng, S.; Feirt, N.; Goldstein, M.; Guarrera, J.; Ippagunta, N.; Ekong, U.; Dun, H.; Lu, Y.; Qu, W.; Schmidt, A.M.; et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004, 39, 422–432. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Muench, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Abdelazeem, K.N.M.; Kalo, M.Z.; Beer-Hammer, S.; Lang, F. The gut microbiota metabolite urolithin A inhibits NF-κB activation in LPS stimulated BMDMs. Sci. Rep. 2021, 11, 7117. [Google Scholar] [CrossRef]
- Visco, D.B.; Manhães de Castro, R.; Guzman-Quevedo, O.; Toscano, A.E. Could polyphenols be used as a neuroprotector therapeutic agent in perinatal brain disorders? Nutr. Neurosci. 2022, 25, 2458–2460. [Google Scholar] [CrossRef]
- Meshalkina, D.; Tsvetkova, E.; Orlova, A.; Islamova, R.; Kysil, E.; Grashina, M.; Gorbach, D.; Babakov, V.; Francioso, A.; Mosca, L.; et al. Neuroprotective and antibacterial effects of phlorotannins isolated from the cell walls of brown algae Fucus vesiculosus and Pelvetia canaliculata. ChemRxiv 2022, 1–30. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s disease and in the gut-brain axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef]
- Guerin, T.; Waterlot, C.; Lipka, E.; Gervois, P.; Bulteel, D.; Betrancourt, D.; Moignard, C.; Nica, A.S.; Furman, C.; Ghinet, A. Ecocatalysed Hurtley reaction synthesis of urolithin derivatives as new potential RAGE antagonists with anti-ageing properties. Sustainable Chem. Pharm. 2021, 23, 100518. [Google Scholar] [CrossRef]
- Godyn, J.; Jonczyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Moulahoum, H.; Ghorbanizamani, F.; Khiari, Z.; Toumi, M.; Benazzoug, Y.; Tok, K.; Timur, S.; Zihnioglu, F. Artemisia alleviates AGE-induced liver complications via MAPK and RAGE signaling pathways modulation: A combinatorial study. Mol. Cell. Biochem. 2022, 477, 2345–2357. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Ho, C.-T.; Li, S. Management of Maillard reaction-derived reactive carbonyl species and advanced glycation end products by tea and tea polyphenols. Food Sci. Hum. Wellness 2022, 11, 557–567. [Google Scholar] [CrossRef]
- Liu, J.; Ye, T.; Zhang, Y.; Zhang, R.; Kong, Y.; Zhang, Y.; Sun, J. Protective Effect of Ginkgolide B against Cognitive Impairment in Mice via Regulation of Gut Microbiota. J. Agric. Food Chem. 2021, 69, 12230–12240. [Google Scholar] [CrossRef]
- Ramya, R.; Coral, K.; Bharathidevi, S.R. RAGE silencing deters CML-AGE induced inflammation and TLR4 expression in endothelial cells. Exp. Eye Res. 2021, 206, 108519. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [PubMed]
- Yatime, L.; Andersen, G.R. Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J. 2013, 280, 6556–6568. [Google Scholar] [CrossRef] [PubMed]
- Moysa, A.; Hammerschmid, D.; Szczepanowski, R.H.; Sobott, F.; Dadlez, M. Enhanced oligomerization of full-length RAGE by synergy of the interaction of its domains. Sci. Rep. 2019, 9, 20332. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.-I.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.-H. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Yan, S.F.; Schmidt, A.M.; Shekhtman, A. Signal Transduction in Receptor for Advanced Glycation End Products (RAGE): Solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem. 2012, 287, 5133–5144. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vetizou, M.; Daillere, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef]
- Wang, J.; Cai, W.; Yu, J.; Liu, H.; He, S.; Zhu, L.; Xu, J. Dietary Advanced Glycation End Products Shift the Gut Microbiota Composition and Induce Insulin Resistance in Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 427–437. [Google Scholar] [CrossRef]
- Bull, M.J.; Plummer, N.T. Part 1: The Human Gut Microbiome in Health and Disease. Integr. Med. 2014, 13, 17–22. [Google Scholar]
- Schaechtle, M.A.; Rosshart, S.P. The microbiota-gut-brain axis in health and disease and its implications for translational research. Front. Cell. Neurosci. 2021, 15, 698172. [Google Scholar] [CrossRef]
- Li, Y.; Ning, L.; Yin, Y.; Wang, R.; Zhang, Z.; Hao, L.; Wang, B.; Zhao, X.; Yang, X.; Yin, L.; et al. Age-related shifts in gut microbiota contribute to cognitive decline in aged rats. Aging 2020, 12, 7801–7817. [Google Scholar] [CrossRef]
- Snelson, M.; Lucut, E.; Coughlan, M.T. The Role of AGE-RAGE Signalling as a Modulator of Gut Permeability in Diabetes. Int. J. Mol. Sci. 2022, 23, 1766. [Google Scholar] [CrossRef]
- Sappington, P.L.; Yang, R.; Yang, H.; Tracey, K.J.; Delude, R.L.; Fink, M.P. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 2002, 123, 790–802. [Google Scholar] [CrossRef]
- Zen, K.; Chen, C.X.J.; Chen, Y.-T.; Wilton, R.; Liu, Y. Receptor for Advanced Glycation Endproducts Mediates Neutrophil Migration across Intestinal Epithelium. J. Immunol. 2007, 178, 2483–2490. [Google Scholar] [CrossRef]
- Weng, M.-H.; Chen, S.-Y.; Li, Z.-Y.; Yen, G.-C. Camellia oil alleviates the progression of Alzheimer’s disease in aluminum chloride-treated rats. Free Radical Biol. Med. 2020, 152, 411–421. [Google Scholar] [CrossRef]
- Moreira, A.P.; Vizuete, A.F.K.; Zin, L.E.F.; de Marques, C.O.; Pacheco, R.F.; Leal, M.B.; Goncalves, C.-A. The Methylglyoxal/RAGE/NOX-2 Pathway is Persistently Activated in the Hippocampus of Rats with STZ-Induced Sporadic Alzheimer Disease. Neurotoxic. Res. 2022, 40, 395–409. [Google Scholar] [CrossRef]
- Wei, C.-C.; Li, S.-W.; Wu, C.-T.; How, C.M.; Pan, M.-H. Dietary Methylglyoxal Exposure Induces Alzheimer’s Disease by Promoting Amyloid β Accumulation and Disrupting Autophagy in Caenorhabditis elegans. J. Agric. Food Chem. 2022, 70, 10011–10021. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Multi-Target Effects of ss-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants 2022, 11, 1199. [Google Scholar] [CrossRef]
- Mirza, F.J.; Zahid, S.; Holsinger, R.M.D. Neuroprotective Effects of Carnosic Acid: Insight into Its Mechanisms of Action. Molecules 2023, 28, 2306. [Google Scholar] [CrossRef]
- Fang, Y.; Doyle, M.F.; Chen, J.; Alosco, M.L.; Mez, J.; Satizabal, C.L.; Qiu, W.Q.; Murabito, J.M.; Lunetta, K.L. Association between inflammatory biomarkers and cognitive aging. PLoS ONE 2022, 17, e0274350. [Google Scholar] [CrossRef]
- Han, Y.; Chen, R.; Lin, Q.; Liu, Y.; Ge, W.; Cao, H.; Li, J. Curcumin improves memory deficits by inhibiting HMGB1-RAGE/TLR4-NF-κB signaling pathway in APPswe/PS1dE9 transgenic mice hippocampus. J. Cell. Mol. Med. 2021, 25, 8947–8956. [Google Scholar] [CrossRef]
- Xue, J.; Ray, R.; Singer, D.; Bohme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Cary, B.P.; Brooks, A.F.; Fawaz, M.V.; Drake, L.R.; Desmond, T.J.; Sherman, P.; Quesada, C.A.; Scott, P.J.H. Synthesis and Evaluation of [18F]RAGER: A First Generation Small-Molecule PET Radioligand Targeting the Receptor for Advanced Glycation Endproducts. ACS Chem. Neurosci. 2016, 7, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Dolores Giron-Gonzalez, M.; Morales-Portillo, A.; Salinas-Castillo, A.; Lopez-Jaramillo, F.J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F.; Salto-Gonzalez, R. Engineered Glycated Amino Dendritic Polymers as Specific Nonviral Gene Delivery Vectors Targeting the Receptor for Advanced Glycation End Products. Bioconjugate Chem. 2014, 25, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Xu, L.; Wang, H.; Yan, X.; Xue, J.; Liu, F.; Hu, J.-F. Atorvastatin exerts its anti-atherosclerotic effects by targeting the receptor for advanced glycation end products. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1130–1137. [Google Scholar] [CrossRef]
- Jyoti, F. Development of New Antibody Based Theranostic Agents Targeting the Receptor for Advanced Glycation End-Product. Ph.D. Thesis, North Dakota State University, Fargo, ND, USA, 2012. [Google Scholar]
- Le Bagge, S.; Fotheringham, A.K.; Leung, S.S.; Forbes, J.M. Targeting the receptor for advanced glycation end products (RAGE) in type 1 diabetes. Med. Res. Rev. 2020, 40, 1200–1219. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.-R.; Liu, N.-F.; Zhu, N.-X. Specific siRNA targeting the receptor for advanced glycation end products inhibits experimental hepatic fibrosis in rats. Int. J. Mol. Sci. 2008, 9, 638–661. [Google Scholar] [CrossRef]
- Zheng, L.; Li, H.; Zhang, W.-S. Recent progress of small-molecule inhibitors targeting the receptor for advanced glycation endproducts. Zhongguo Yaowu Huaxue Zazhi 2021, 31, 68–77. [Google Scholar]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- Sanajou, D.; Ghorbani Haghjo, A.; Argani, H.; Aslani, S. AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef]
- Ren, L.; Yan, H. Targeting AGEs-RAGE pathway inhibits inflammation and presents neuroprotective effect against hepatic ischemia-reperfusion induced hippocampus damage. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101792. [Google Scholar] [CrossRef]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Oshitari, T. Advanced Glycation End-Products and Diabetic Neuropathy of the Retina. Int. J. Mol. Sci. 2023, 24, 2927. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Rabbani, P.; Egana-Gorrono, L.; Quadri, N.; Frye, L.; Zhou, B.; Reverdatto, S.; Ramirez, L.S.; Dansereau, S.; Pan, J.; et al. Small-molecule antagonism of the interaction of the RAGE cytoplasmic domain with DIAPH1 reduces diabetic complications in mice. Sci. Transl. Med. 2021, 13, eabf7084. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, V.P.; Aryal, P.; Soni, P. RAGE Inhibitors in Neurodegenerative Diseases. Biomedicines 2023, 11, 1131. https://doi.org/10.3390/biomedicines11041131
Reddy VP, Aryal P, Soni P. RAGE Inhibitors in Neurodegenerative Diseases. Biomedicines. 2023; 11(4):1131. https://doi.org/10.3390/biomedicines11041131
Chicago/Turabian StyleReddy, V. Prakash, Puspa Aryal, and Pallavi Soni. 2023. "RAGE Inhibitors in Neurodegenerative Diseases" Biomedicines 11, no. 4: 1131. https://doi.org/10.3390/biomedicines11041131
APA StyleReddy, V. P., Aryal, P., & Soni, P. (2023). RAGE Inhibitors in Neurodegenerative Diseases. Biomedicines, 11(4), 1131. https://doi.org/10.3390/biomedicines11041131