
Epigenetics Approaches toward Precision Medicine for Idiopathic Pulmonary Fibrosis: Focus on DNA Methylation

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epigenetics
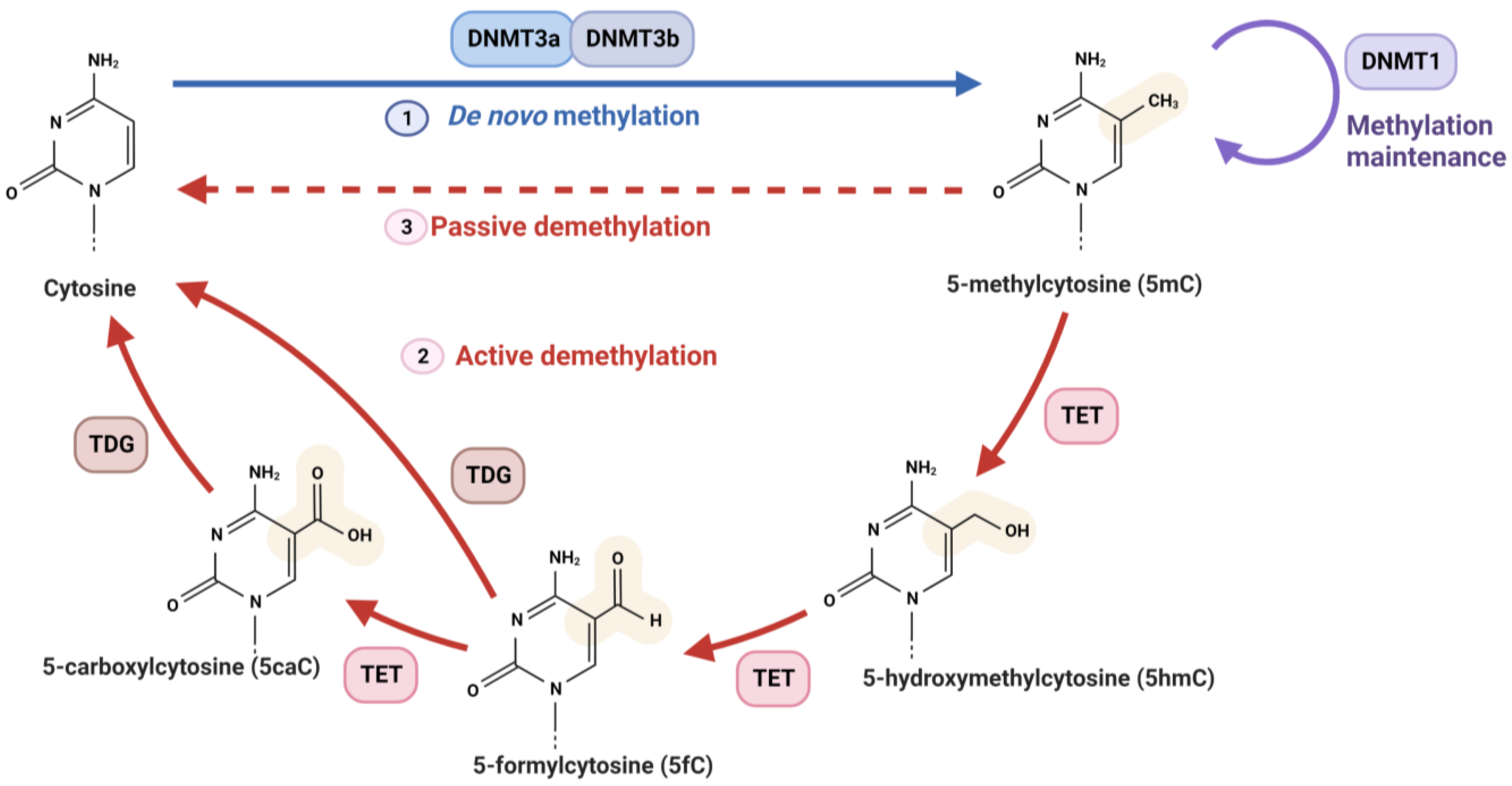
2.1. DNA Methylation
2.1.1. DNMTs
2.1.2. MBPs
2.1.3. TETs
3. DNA Methylation in Fibrosis
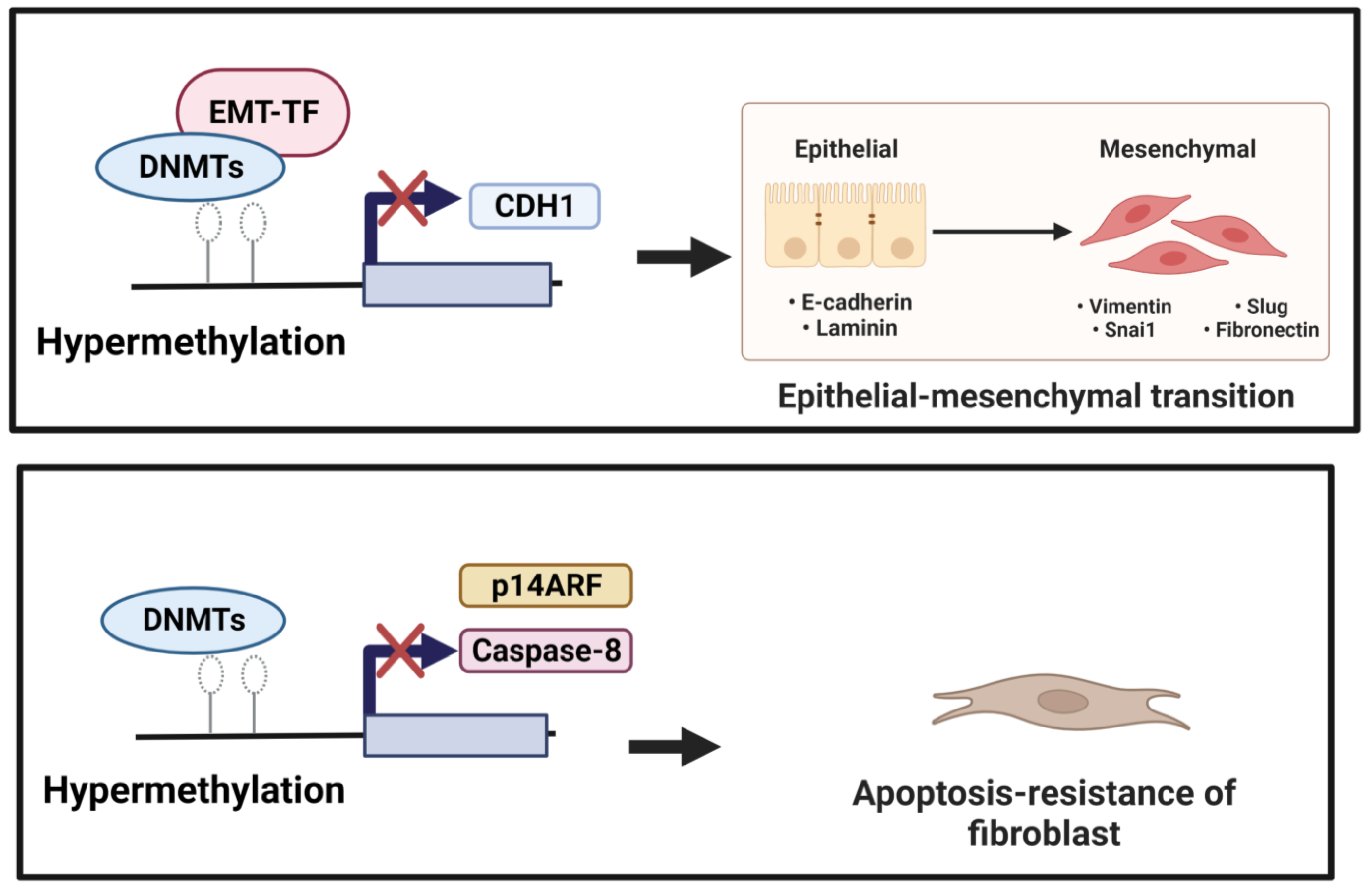
3.1. DNA Methylation and EMT
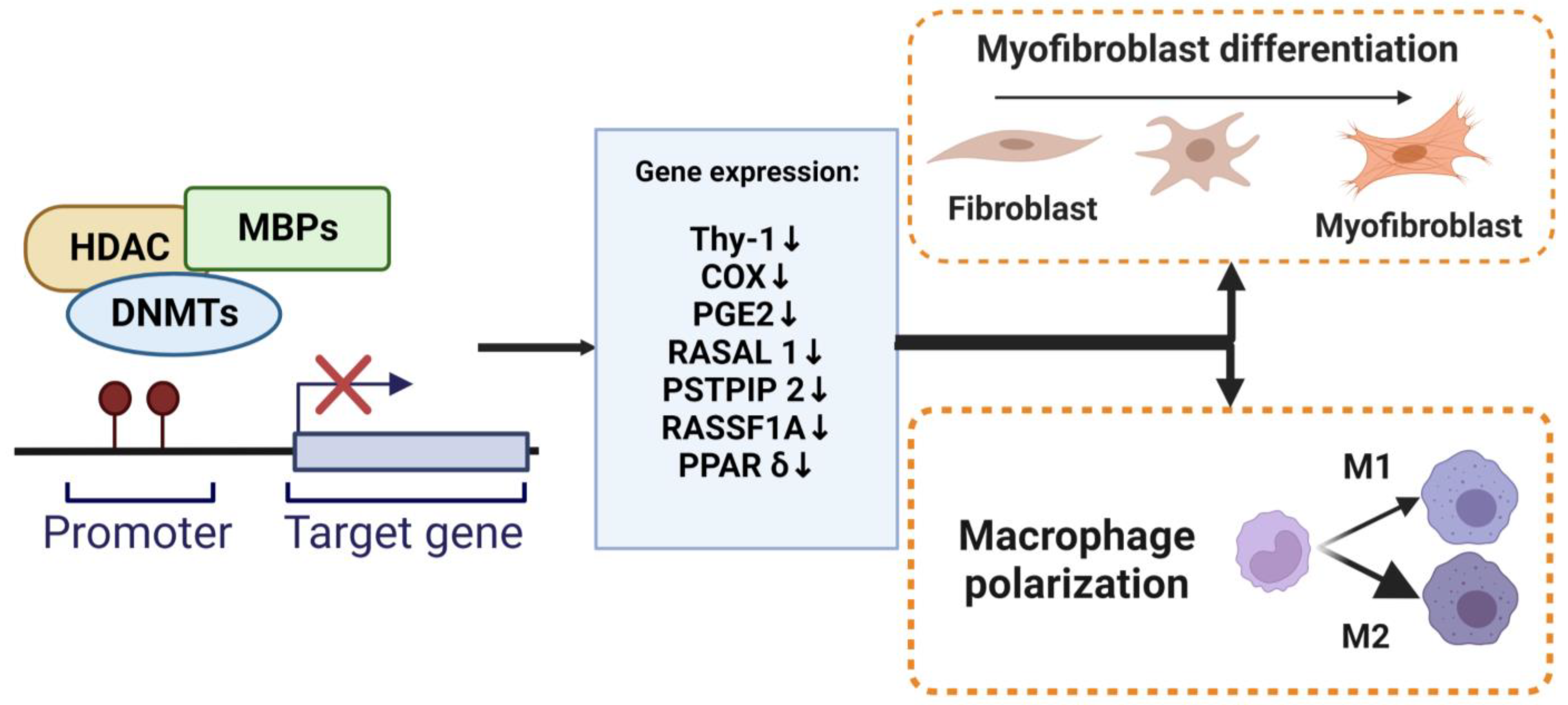
3.2. DNA Methylation and Myofibroblast Differentiation
3.3. DNA Methylation and Macrophage Polarization
4. Targeted Gene and Personalized Medicine
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 148. [Google Scholar] [CrossRef]
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457. [Google Scholar] [CrossRef] [PubMed]
- Fernández Fabrellas, E.; Peris Sánchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Alqalyoobi, S.; Fernández Pérez, E.R.; Oldham, J.M. In-hospital mortality trends among patients with idiopathic pulmonary fibrosis in the United States between 2013–2017: A comparison of academic and non-academic programs. BMC Pulm. Med. 2020, 20, 289. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-W.; Kim, J.; Song, J.W. Impact of the revised definition on incidence and outcomes of acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2022, 12, 8817. [Google Scholar] [CrossRef] [PubMed]
- Aburto, M.; Herráez, I.; Iturbe, D.; Jiménez-Romero, A. Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis. Med. Sci. 2018, 6, 73. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Paulsen, M.; Toth, R.; Weichenhan, D.; Butz, S.; Schatterny, J.; Liebers, R.; Lutsik, P.; Plass, C.; Mall, M.A. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat. Commun. 2021, 12, 6520. [Google Scholar] [CrossRef] [PubMed]
- Horgan, R.P.; Kenny, L.C. ‘Omic’ technologies: Genomics, transcriptomics, proteomics and metabolomics. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Kan, M.; Shumyatcher, M.; Himes, B.E. Using omics approaches to understand pulmonary diseases. Respir. Res. 2017, 18, 149. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef]
- Guiot, J.; Struman, I.; Chavez, V.; Henket, M.; Herzog, M.; Scoubeau, K.; Hardat, N.; Bondue, B.; Corhay, J.L.; Moermans, C.; et al. Altered epigenetic features in circulating nucleosomes in idiopathic pulmonary fibrosis. Clin. Epigenetics 2017, 9, 84. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 1546. [Google Scholar] [CrossRef]
- Mehrmohamadi, M.; Sepehri, M.H.; Nazer, N.; Norouzi, M.R. A Comparative Overview of Epigenomic Profiling Methods. Front. Cell Dev. Biol. 2021, 9, 714687. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Naseer, M.I.; Manan, A.; Ansari, S.A.; Begum, I.; Qazi, M.H.; Pushparaj, P.N.; Abuzenadah, A.M.; Al-Qahtani, M.H.; et al. The role of epigenetics in personalized medicine: Challenges and opportunities. BMC Med. Genomics 2015, 8, S5. [Google Scholar] [CrossRef] [PubMed]
- Magnini, D.; Montemurro, G.; Iovene, B.; Tagliaboschi, L.; Gerardi, R.E.; Lo Greco, E.; Bruni, T.; Fabbrizzi, A.; Lombardi, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Molecular Endotypes of Fibrosis Stratifying Existing and Emerging Therapies. Respiration. 2017, 93, 379–395. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Lacal, I.; Ventura, R. Epigenetic Inheritance: Concepts, Mechanisms and Perspectives. Front. Mol. Neurosci. 2018, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.K.M.; Katz, L.A. Epigenetics as Driver of Adaptation and Diversification in Microbial Eukaryotes. Front. Genet. 2021, 12, 642220. [Google Scholar] [CrossRef]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 2013, 38, 220–236. [Google Scholar] [CrossRef]
- Breton, C.V.; Landon, R.; Kahn, L.G.; Enlow, M.B.; Peterson, A.K.; Bastain, T.; Braun, J.; Comstock, S.S.; Duarte, C.S.; Hipwell, A.; et al. Exploring the evidence for epigenetic regulation of environmental influences on child health across generations. Commun. Biol. 2021, 4, 769. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef]
- Toraño, E.G.; García, M.G.; Fernández-Morera, J.L.; Niño-García, P.; Fernández, A.F. The Impact of External Factors on the Epigenome: In Utero and over Lifetime. Biomed Res. Int. 2016, 2016, 2568635. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Kim, S.; Kaang, B.-K. Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 2017, 49, e281. [Google Scholar] [CrossRef]
- Takatsuka, H.; Umeda, M. Epigenetic Control of Cell Division and Cell Differentiation in the Root Apex. Front. Plant Sci. 2015, 6, 1178. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Venditti, S.; Verdone, L.; Reale, A.; Vetriani, V.; Caserta, M.; Zampieri, M. Molecules of Silence: Effects of Meditation on Gene Expression and Epigenetics. Front. Psychol. 2020, 11, 1767. [Google Scholar] [CrossRef]
- De Sadeleer, L.J.; McDonough, J.E.; Schupp, J.C.; Yan, X.; Vanstapel, A.; Van Herck, A.; Everaerts, S.; Geudens, V.; Sacreas, A.; Goos, T.; et al. Lung Microenvironments and Disease Progression in Fibrotic Hypersensitivity Pneumonitis. Am. J. Respir. Crit. Care Med. 2022, 205, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Wang, S.; Duleba, M.; Niroula, S.; Goller, K.; Xie, J.; Mahalingam, R.; Neupane, R.; Liew, A.-A.; Vincent, M.; et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 2020, 181, 848–864.e18. [Google Scholar] [CrossRef] [PubMed]
- McErlean, P.; Bell, C.G.; Hewitt, R.J.; Busharat, Z.; Ogger, P.P.; Ghai, P.; Albers, G.J.; Calamita, E.; Kingston, S.; Molyneaux, P.L.; et al. DNA Methylome Alterations Are Associated with Airway Macrophage Differentiation and Phenotype during Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Maremanda, K.P.; Campos, M.; Chand, H.S.; Li, F.; Hirani, N.; Haseeb, M.A.; Li, D.; Rahman, I. Distinct Exosomal miRNA Profiles from BALF and Lung Tissue of COPD and IPF Patients. Int. J. Mol. Sci. 2021, 22, 11830. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, P.; Ke, S.; Dong, H.; Zhan, M.; Hu, Q.; Li, J. Histone methyltransferase SETDB1 inhibits TGF-β-induced epithelial-mesenchymal transition in pulmonary fibrosis by regulating SNAI1 expression and the ferroptosis signaling pathway. Arch. Biochem. Biophys. 2022, 715, 109087. [Google Scholar] [CrossRef]
- Morales-Nebreda, L.; McLafferty, F.S.; Singer, B.D. DNA methylation as a transcriptional regulator of the immune system. Transl. Res. 2019, 204, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, X.; Liu, Q.; Zhao, Y.; Jiang, W.; Wu, A.; Zhou, D.-X. DNA demethylases remodel DNA methylation in rice gametes and zygote and are required for reproduction. Mol. Plant 2021, 14, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Bommarito, P.A.; Fry, R.C. Chapter 2-1—The Role of DNA Methylation in Gene Regulation. In Toxicoepigenetics; McCullough, S.D., Dolinoy, D.C., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 127–151. ISBN 978-0-12-812433-8. [Google Scholar]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin 2021, 14, 25. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef]
- Luo, Q.-K.; Zhang, H.; Li, L. Research Advances on DNA Methylation in Idiopathic Pulmonary Fibrosis. In Single-Cell Sequencing and Methylation: Methods and Clinical Applications; Yu, B., Zhang, J., Zeng, Y., Li, L., Wang, X., Eds.; Springer: Singapore, 2020; pp. 73–81. ISBN 978-981-15-4494-1. [Google Scholar]
- Xiao, F.-H.; Wang, H.-T.; Kong, Q.-P. Dynamic DNA Methylation During Aging: A “Prophet” of Age-Related Outcomes. Front. Genet. 2019, 10, 107. [Google Scholar] [CrossRef]
- Chakraborty, A.; Viswanathan, P. Methylation-Demethylation Dynamics: Implications of Changes in Acute Kidney Injury. Anal. Cell. Pathol. 2018, 2018, 8764384. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Kostrzewa, R.M.; Filip, M. A Comprehensive View of the Epigenetic Landscape Part I: DNA Methylation, Passive and Active DNA Demethylation Pathways and Histone Variants. Neurotox. Res. 2015, 27, 84–97. [Google Scholar] [CrossRef]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef]
- Fournier, A.; Sasai, N.; Nakao, M.; Defossez, P.-A. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief. Funct. Genomics 2011, 11, 251–264. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Onodera, A.; González-Avalos, E.; Lio, C.-W.J.; Georges, R.O.; Bellacosa, A.; Nakayama, T.; Rao, A. Roles of TET and TDG in DNA demethylation in proliferating and non-proliferating immune cells. Genome Biol. 2021, 22, 186. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sun, H.; Lin, L.; Zhang, Y.; Chen, J.; Liang, L.; Li, Y.; Zhang, M.; Yang, X.; Wang, X.; et al. Passive DNA demethylation preferentially up-regulates pluripotency-related genes and facilitates the generation of induced pluripotent stem cells. J. Biol. Chem. 2017, 292, 18542–18555. [Google Scholar] [CrossRef] [PubMed]
- Ohno, R.; Nakayama, M.; Naruse, C.; Okashita, N.; Takano, O.; Tachibana, M.; Asano, M.; Saitou, M.; Seki, Y. A replication-dependent passive mechanism modulates DNA demethylation in mouse primordial germ cells. Development 2013, 140, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wu, L.; Luo, Z.; Zhang, M.; Lai, J.; Li, L.; Springer, N.M.; Li, Q. DNA demethylation affects imprinted gene expression in maize endosperm. Genome Biol. 2022, 23, 77. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, M.; Lyu, X.; Li, C.; Thannickal, V.J.; Sanders, Y.Y. DNA methylation regulated gene expression in organ fibrosis. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2389–2397. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Bartczak, K.; Białas, A.J.; Kotecki, M.J.; Górski, P.; Piotrowski, W.J. More than a Genetic Code: Epigenetics of Lung Fibrosis. Mol. Diagnosis Ther. 2020, 24, 665–681. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Alhosin, M.; Hamiche, A.; Mousli, M. Coordinated Dialogue between UHRF1 and DNMT1 to Ensure Faithful Inheritance of Methylated DNA Patterns. Genes 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef]
- Gao, L.; Emperle, M.; Guo, Y.; Grimm, S.A.; Ren, W.; Adam, S.; Uryu, H.; Zhang, Z.-M.; Chen, D.; Yin, J.; et al. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 2020, 11, 3355. [Google Scholar] [CrossRef]
- Giehr, P.; Kyriakopoulos, C.; Ficz, G.; Wolf, V.; Walter, J. The Influence of Hydroxylation on Maintaining CpG Methylation Patterns: A Hidden Markov Model Approach. PLOS Comput. Biol. 2016, 12, e1004905. [Google Scholar] [CrossRef]
- Tao, H.; Shi, P.; Zhao, X.-D.; Xuan, H.-Y.; Gong, W.-H.; Ding, X.-S. DNMT1 deregulation of SOCS3 axis drives cardiac fibroblast activation in diabetic cardiac fibrosis. J. Cell. Physiol. 2021, 236, 3481–3494. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β–induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef]
- Wei, A.; Gao, Q.; Chen, F.; Zhu, X.; Chen, X.; Zhang, L.; Su, X.; Dai, J.; Shi, Y.; Cao, W. Inhibition of DNA methylation de-represses peroxisome proliferator-activated receptor-γ and attenuates pulmonary fibrosis. Br. J. Pharmacol. 2022, 179, 1304–1318. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: A historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Ginder, G.D.; Williams, D.C.J. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Ther. 2018, 184, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic factors MeCP2 and HDAC6 control α-tubulin acetylation in cardiac fibroblast proliferation and fibrosis. Inflamm. Res. 2016, 65, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wu, G.-R.; Wang, Q.; Pan, T.; Zhang, L.; Xu, Y.; Xiong, W.; Zhou, Q.; Wang, Y. Macrophage-targeted delivery of siRNA to silence Mecp2 gene expression attenuates pulmonary fibrosis. Bioeng. Transl. Med. 2022, 7, e10280. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, X.; Zhang, R.; Cao, T.; Cai, S.-Y.; Boyer, J.L.; Zhang, X.; Li, D.; Huang, Y. A Positive Feedback Loop of TET3 and TGF-β1 Promotes Liver Fibrosis. Cell Rep. 2020, 30, 1310–1318. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, J.; Zhang, H.; Shen, Z.; Liu, H.; Lv, S.; Yu, X.; Zhang, D.; Ding, X.; Zhang, X. Hydrogen sulfide attenuates renal fibrosis by inducing TET-dependent DNA demethylation on Klotho promoter. FASEB J. 2020, 34, 11474–11487. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Wan, P.; Long, E.; Li, Z.; Zhu, Y.; Su, W.; Zhuo, Y. TET-dependent GDF7 hypomethylation impairs aqueous humor outflow and serves as a potential therapeutic target in glaucoma. Mol. Ther. 2021, 29, 1639–1657. [Google Scholar] [CrossRef]
- Maamari, S.; Xu, X.; Zeisberg, E. Demethylation and reactivation of the fibrosis-suppressor gene Rasal1 is initiated by R-loop formation which facilitates recruitment of TET3 and GADD45g. Eur. Heart J. 2022, 43, ehac544-2879. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung Fibroblasts from Patients with Idiopathic Pulmonary Fibrosis Exhibit Genome-Wide Differences in DNA Methylation Compared to Fibroblasts from Nonfibrotic Lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef]
- Lee, J.-U.; Son, J.-H.; Shim, E.-Y.; Cheong, H.S.; Shin, S.-W.; Shin, H.D.; Baek, A.R.; Ryu, S.; Park, C.-S.; Chang, H.S.; et al. Global DNA Methylation Pattern of Fibroblasts in Idiopathic Pulmonary Fibrosis. DNA Cell Biol. 2019, 38, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global Methylation Patterns in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef]
- Wielscher, M.; Vierlinger, K.; Kegler, U.; Ziesche, R.; Gsur, A.; Weinhäusel, A. Diagnostic Performance of Plasma DNA Methylation Profiles in Lung Cancer, Pulmonary Fibrosis and COPD. EBioMedicine 2015, 2, 929–936. [Google Scholar] [CrossRef]
- Hata, A.; Nakajima, T.; Matsusaka, K.; Fukuyo, M.; Morimoto, J.; Yamamoto, T.; Sakairi, Y.; Rahmutulla, B.; Ota, S.; Wada, H.; et al. A low DNA methylation epigenotype in lung squamous cell carcinoma and its association with idiopathic pulmonary fibrosis and poorer prognosis. Int. J. Cancer 2020, 146, 388–399. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.; van der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; van Duijn, C.M.; Brandsma, C.-A.; van Diemen, C.C.; Vonk, J.M.; et al. From blood to lung tissue: Effect of cigarette smoke on DNA methylation and lung function. Respir. Res. 2018, 19, 212. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Urbanova, M.; Buocikova, V.; Trnkova, L.; Strapcova, S.; Kajabova, V.H.; Melian, E.B.; Novisedlakova, M.; Tomas, M.; Dubovan, P.; Earl, J.; et al. DNA Methylation Mediates EMT Gene Expression in Human Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 2117. [Google Scholar] [CrossRef]
- Park, J.-H.; Shin, J.-M.; Yang, H.-W.; Park, I.-H. DNMTs Are Involved in TGF-β1-Induced Epithelial-Mesenchymal Transitions in Airway Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 3003. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells 2013, 2, 545–573. [Google Scholar] [CrossRef]
- Kostova, I.; Mandal, R.; Becker, S.; Strebhardt, K. The role of caspase-8 in the tumor microenvironment of ovarian cancer. Cancer Metastasis Rev. 2021, 40, 303–318. [Google Scholar] [CrossRef]
- Cisneros, J.; Hagood, J.; Checa, M.; Ortiz-Quintero, B.; Negreros, M.; Herrera, I.; Ramos, C.; Pardo, A.; Selman, M. Hypermethylation-mediated silencing of p14(ARF) in fibroblasts from idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L295–L303. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wei, X.; Ye, T.; Hang, L.; Mou, L.; Su, J. Interrelation Between Fibroblasts and T Cells in Fibrosing Interstitial Lung Diseases. Front. Immunol. 2021, 12, 747335. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast–Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 1459. [Google Scholar] [CrossRef]
- Doolin, M.T.; Smith, I.M.; Stroka, K.M. Fibroblast to myofibroblast transition is enhanced by increased cell density. Mol. Biol. Cell 2021, 32, ar41. [Google Scholar] [CrossRef]
- Li, J.; Yao, W.; Zhang, L.; Bao, L.; Chen, H.; Wang, D.; Yue, Z.; Li, Y.; Zhang, M.; Hao, C. Genome-wide DNA methylation analysis in lung fibroblasts co-cultured with silica-exposed alveolar macrophages. Respir. Res. 2017, 18, 91. [Google Scholar] [CrossRef]
- He, Y.; Ling, S.; Sun, Y.; Sheng, Z.; Chen, Z.; Pan, X.; Ma, G. DNA methylation regulates α-smooth muscle actin expression during cardiac fibroblast differentiation. J. Cell. Physiol. 2019, 234, 7174–7185. [Google Scholar] [CrossRef]
- Neveu, W.A.; Mills, S.T.; Staitieh, B.S.; Sueblinvong, V. TGF-β1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am. J. Physiol. Cell Physiol. 2015, 309, C616–C626. [Google Scholar] [CrossRef]
- Ogawa, H.; Koyanagi-Aoi, M.; Otani, K.; Zen, Y.; Maniwa, Y.; Aoi, T. Interleukin-6 blockade attenuates lung cancer tissue construction integrated by cancer stem cells. Sci. Rep. 2017, 7, 12317. [Google Scholar] [CrossRef]
- Al-Kharashi, L.A.; Al-Mohanna, F.H.; Tulbah, A.; Aboussekhra, A. The DNA methyl-transferase protein DNMT1 enhances tumor-promoting properties of breast stromal fibroblasts. Oncotarget 2018, 9, 2329–2343. [Google Scholar] [CrossRef]
- Evans, I.C.; Barnes, J.L.; Garner, I.M.; Pearce, D.R.; Maher, T.M.; Shiwen, X.; Renzoni, E.A.; Wells, A.U.; Denton, C.P.; Laurent, G.J.; et al. Epigenetic regulation of cyclooxygenase-2 by methylation of c8orf4 in pulmonary fibrosis. Clin. Sci. 2016, 130, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Oakley, F.; Akiboye, F.; Elsharkawy, A.; Thorne, A.W.; Mann, D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 2007, 14, 275–285. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhou, Q.; Hu, M.; Sanders, Y.Y. MeCP2 epigenetically regulates alpha-smooth muscle actin in human lung fibroblasts. J. Cell. Biochem. 2020, 121, 3616–3625. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-M.; Um, J.-Y.; Lee, S.-A.; Park, I.-H.; Lee, S.-H.; Lee, H.-M. Effect of MeCP2 on TGF-β1-induced Extracellular Matrix Production in Nasal Polyp-derived Fibroblasts. Am. J. Rhinol. Allergy 2018, 32, 228–235. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Wu, G.-R.; Zhou, Q.; Yue, H.; Rao, L.-Z.; Yuan, T.; Mo, B.; Wang, F.-X.; Chen, L.-M.; et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci. Adv. 2021, 7, eabb6075. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Huang, T.; Wu, G.-R.; Zhou, Q.; Wang, F.-X.; Chen, L.-M.; Sun, F.; Lv, Y.; Xiong, F.; et al. The methyl-CpG-binding domain 2 facilitates pulmonary fibrosis by orchestrating fibroblast to myofibroblast differentiation. Eur. Respir. J. 2022, 60, 2003697. [Google Scholar] [CrossRef]
- He, Y.; Tsou, P.-S.; Khanna, D.; Sawalha, A.H. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann. Rheum. Dis. 2018, 77, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef]
- Robinson, C.M.; Neary, R.; Levendale, A.; Watson, C.J.; Baugh, J.A. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 2012, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, F.; Irnaten, M.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Hypoxia-Induced Changes in DNA Methylation Alter RASAL1 and TGFβ1 Expression in Human Trabecular Meshwork Cells. PLoS ONE 2016, 11, e0153354. [Google Scholar] [CrossRef] [PubMed]
- Rajgarhia, A.; Ayasolla, K.R.; Zaghloul, N.; Lopez Da Re, J.M.; Miller, E.J.; Ahmed, M. Extracellular Superoxide Dismutase (EC-SOD) Regulates Gene Methylation and Cardiac Fibrosis During Chronic Hypoxic Stress. Front. Cardiovasc. Med. 2021, 8, 669975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Chen, Y.; Armstrong, D.A.; Salas, L.A.; Hazlett, H.F.; Nymon, A.B.; Dessaint, J.A.; Aridgides, D.S.; Mellinger, D.L.; Liu, X.; Christensen, B.C.; et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenetics 2018, 10, 152. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, X.; Li, W.; Huang, H.; Li, H.; Pan, X.; Li, X.; Huang, C.; Meng, X.; Zhang, L.; et al. PSTPIP2 connects DNA methylation to macrophage polarization in CCL4-induced mouse model of hepatic fibrosis. Oncogene 2018, 37, 6119–6135. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef]
- Qin, W.; Spek, C.A.; Scicluna, B.P.; van der Poll, T.; Duitman, J. Myeloid DNA methyltransferase3b deficiency aggravates pulmonary fibrosis by enhancing profibrotic macrophage activation. Respir. Res. 2022, 23, 162. [Google Scholar] [CrossRef]
- Sun, W. The role of MBD2 regulating M1 macrophage polarization in acute lung injury. Eur. Respir. J. 2022, 60, 4589. [Google Scholar] [CrossRef]
- Zhao, D.; Mokhtari, R.; Pedrosa, E.; Birnbaum, R.; Zheng, D.; Lachman, H.M. Transcriptome analysis of microglia in a mouse model of Rett syndrome: Differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol. Autism 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Ai, K.; Pan, J.; Zhang, P.; Li, H.; He, Z.; Zhang, H.; Li, X.; Li, Y.; Yi, L.; Kang, Y.; et al. Methyl-CpG-binding domain protein 2 contributes to renal fibrosis through promoting polarized M1 macrophages. Cell Death Dis. 2022, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.; Snetsinger, B.; Rauh, M.J. Tet2 Is a Novel Regulator of Murine Macrophage Differentiation and Polarization. Blood 2015, 126, 646. [Google Scholar] [CrossRef]
- Hams, E.; Saunders, S.P.; Cummins, E.P.; O’Connor, A.; Tambuwala, M.T.; Gallagher, W.M.; Byrne, A.; Campos-Torres, A.; Moynagh, P.M.; Jobin, C.; et al. The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock 2011, 36, 295–302. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Strianese, O.; Rizzo, F.; Ciccarelli, M.; Galasso, G.; D’Agostino, Y.; Salvati, A.; Del Giudice, C.; Tesorio, P.; Rusciano, M.R. Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease. Genes 2020, 11, 747. [Google Scholar] [CrossRef]
- Priyadharshini, V.S.; Teran, L.M. Personalized Medicine in Respiratory Disease: Role of Proteomics. Adv. Protein Chem. Struct. Biol. 2016, 102, 115–146. [Google Scholar]
- Agustí, A.; Bafadhel, M.; Beasley, R.; Bel, E.H.; Faner, R.; Gibson, P.G.; Louis, R.; McDonald, V.M.; Sterk, P.J.; Thomas, M.; et al. Precision medicine in airway diseases: Moving to clinical practice. Eur. Respir. J. 2017, 50, 1701655. [Google Scholar] [CrossRef]
- Leung, J.M.; Obeidat, M.; Sadatsafavi, M.; Sin, D.D. Introduction to precision medicine in COPD. Eur. Respir. J. 2019, 53, 1802460. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Kaminski, N. Personalized medicine: Applying “omics” to lung fibrosis. Biomark. Med. 2012, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Locy, M.L.; Luckhardt, T.R.; Thannickal, V.J. Targeted Therapy for Idiopathic Pulmonary Fibrosis: Where To Now? Drugs 2016, 76, 291–300. [Google Scholar] [CrossRef]
- Ruigrok, M.J.R.; Frijlink, H.W.; Melgert, B.N.; Olinga, P.; Hinrichs, W.L.J. Gene therapy strategies for idiopathic pulmonary fibrosis: Recent advances, current challenges, and future directions. Mol. Ther. Methods Clin. Dev. 2021, 20, 483–496. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Tsitoura, E.; Vasarmidi, E.; Symvoulakis, E.K.; Aidinis, V.; Tzilas, V.; Tzouvelekis, A.; Bouros, D. Precision medicine in idiopathic pulmonary fibrosis therapy: From translational research to patient-centered care. Curr. Opin. Pharmacol. 2021, 57, 71–80. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, R.-Z.; Ruan, J.; Zhu, X.-B.; Yang, Y. Up-regulation of THY1 attenuates interstitial pulmonary fibrosis and promotes lung fibroblast apoptosis during acute interstitial pneumonia by blockade of the WNT signaling pathway. Cell Cycle 2019, 18, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Miguel, N.; Hagood, J.S.; Espinoza, C.R.; Balderas-Martínez, Y.I.; Selman, M.; Pardo, A. Correction: Transforming growth factor beta 1 induces methylation changes in lung fibroblasts. PLoS ONE 2019, 14, e0224841. [Google Scholar]
- Li, X.; Feng, C.; Peng, S. Epigenetics alternation in lung fibrosis and lung cancer. Front. Cell Dev. Biol. 2022, 10, 1060201. [Google Scholar] [CrossRef]
- Wong, C.C.; Kang, W.; Xu, J.; Qian, Y.; Luk, S.T.Y.; Chen, H.; Li, W.; Zhao, L.; Zhang, X.; Chiu, P.W.; et al. Prostaglandin E(2) induces DNA hypermethylation in gastric cancer in vitro and in vivo. Theranostics 2019, 9, 6256–6268. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Yang, C.-C.; Pan, S.-Y.; Chou, Y.-H.; Chang, F.-C.; Lai, C.-F.; Tsai, M.-H.; Hsu, H.-L.; Lin, C.-H.; Chiang, W.C.; et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effendi, W.I.; Nagano, T. Epigenetics Approaches toward Precision Medicine for Idiopathic Pulmonary Fibrosis: Focus on DNA Methylation. Biomedicines 2023, 11, 1047. https://doi.org/10.3390/biomedicines11041047
Effendi WI, Nagano T. Epigenetics Approaches toward Precision Medicine for Idiopathic Pulmonary Fibrosis: Focus on DNA Methylation. Biomedicines. 2023; 11(4):1047. https://doi.org/10.3390/biomedicines11041047
Chicago/Turabian StyleEffendi, Wiwin Is, and Tatsuya Nagano. 2023. "Epigenetics Approaches toward Precision Medicine for Idiopathic Pulmonary Fibrosis: Focus on DNA Methylation" Biomedicines 11, no. 4: 1047. https://doi.org/10.3390/biomedicines11041047
APA StyleEffendi, W. I., & Nagano, T. (2023). Epigenetics Approaches toward Precision Medicine for Idiopathic Pulmonary Fibrosis: Focus on DNA Methylation. Biomedicines, 11(4), 1047. https://doi.org/10.3390/biomedicines11041047