
Interaction between Gut Microbiota and Dendritic Cells in Colorectal Cancer


Abstract
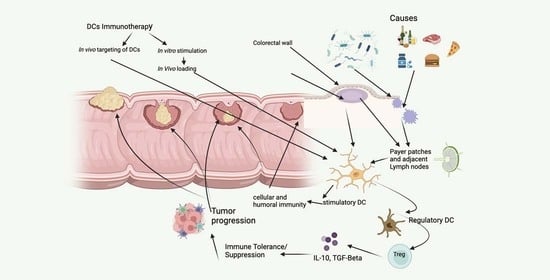
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
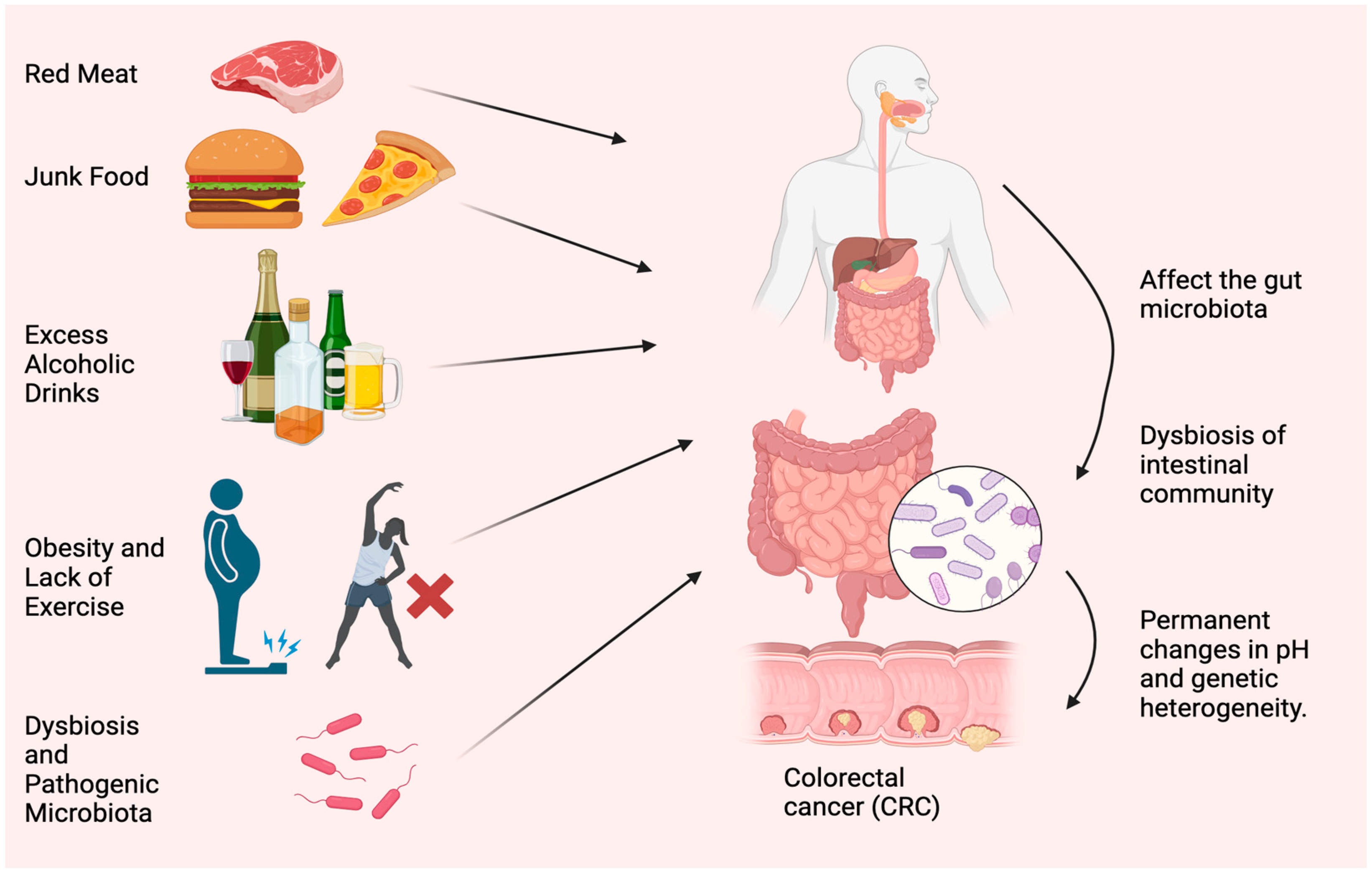
2. Causes and Symptoms of CRC
2.1. Effect of Red Meat and Junk Food
2.2. Correlation between Healthy Diet and CRC
2.3. Role of Obesity and Physical Activity on CRC
2.4. The Role of GUT Microbiota in Dysbiosis and CRC
2.4.1. Fusobacterium nucleatum
2.4.2. Enterotoxigenic Bacteroides fragilis (ETBF)
2.4.3. Escherichia coli (E. coli)
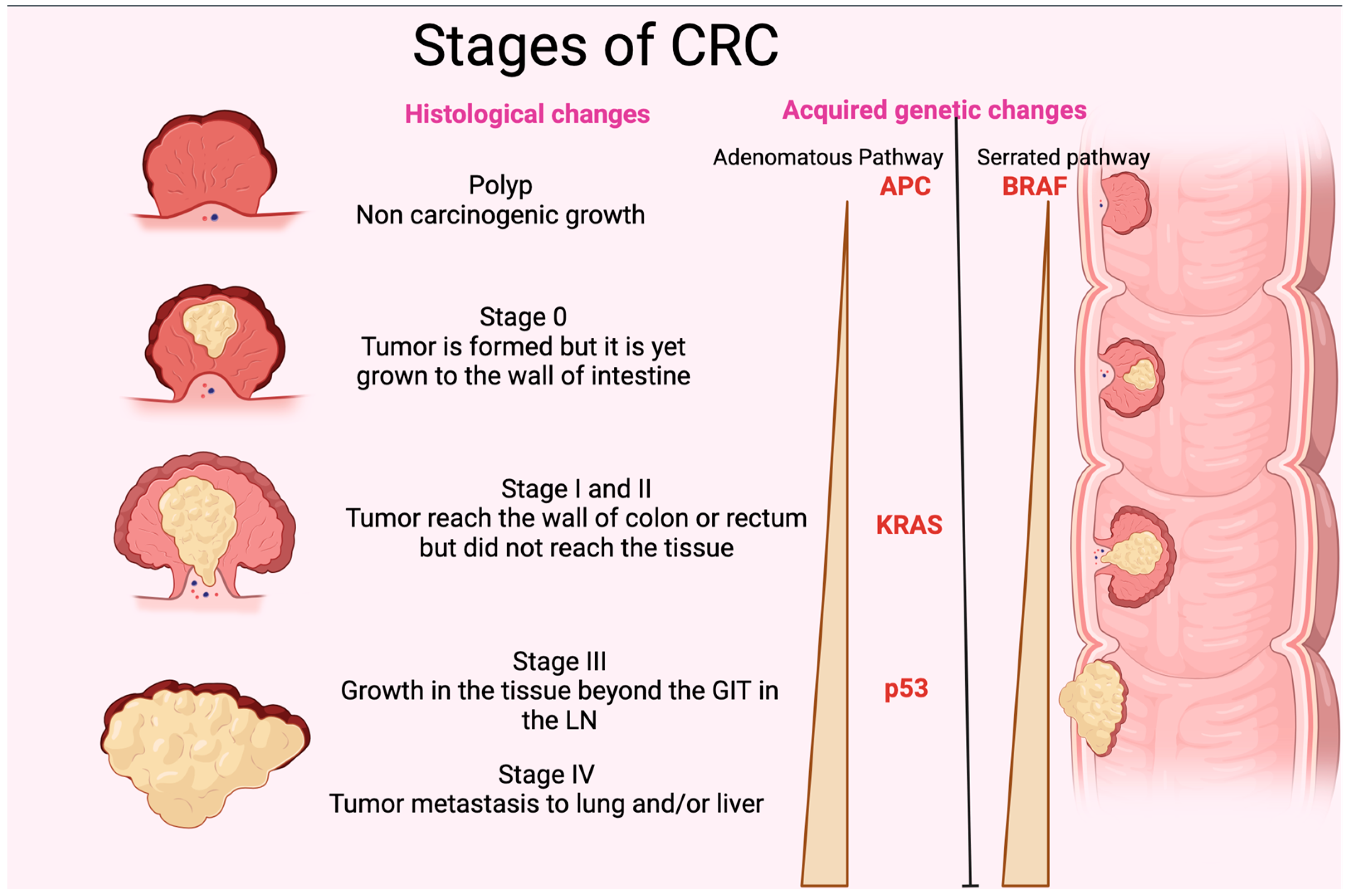
3. Pathogenesis and Genetic Alteration of CRC
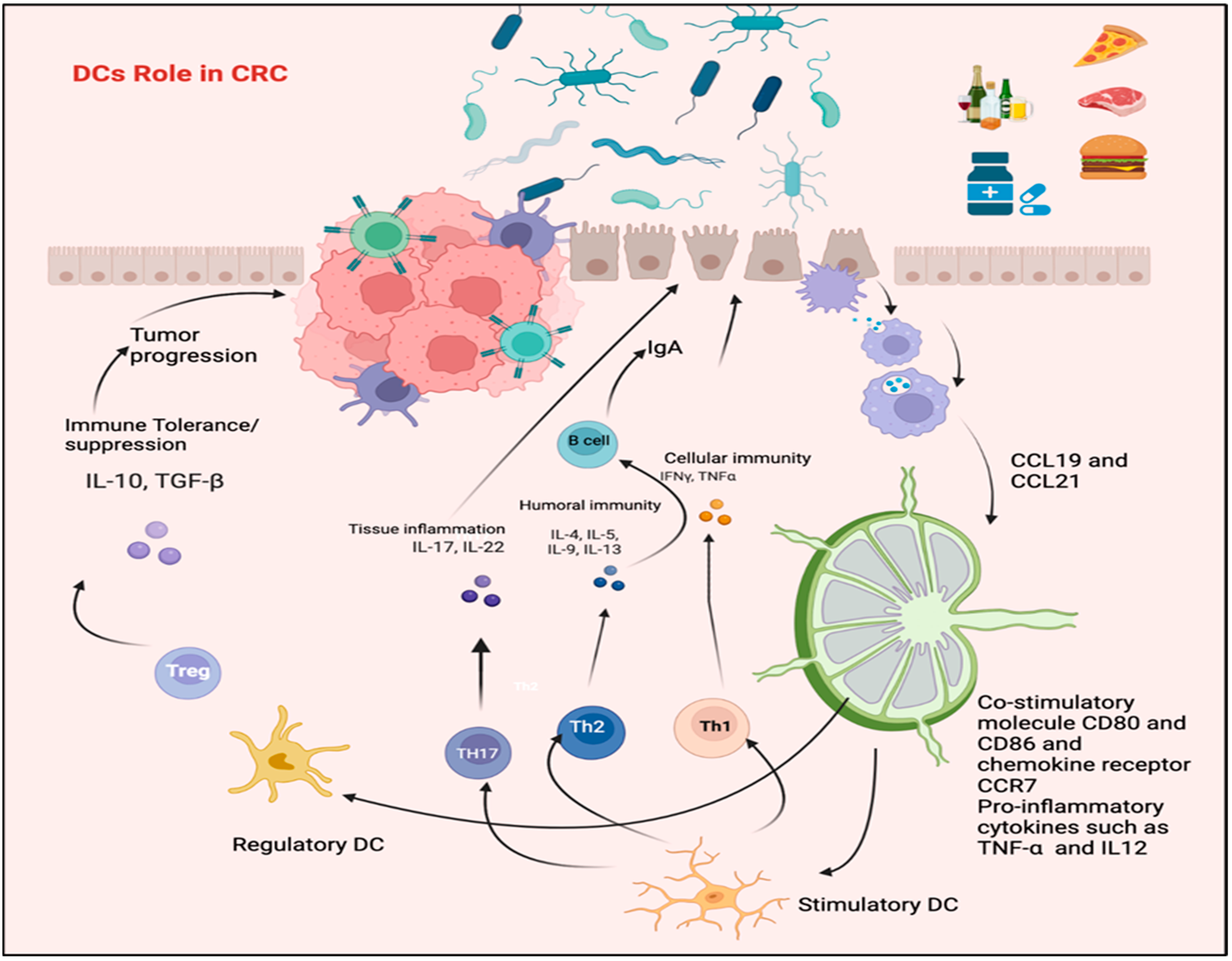
4. Dendritic Cells and the Regulation of Immune Response in CRC
5. Alteration in DCs Phenotypic Profile in CRC Patients
5.1. The Role of T Lymphocyte and IL-17 in CRC
5.2. Role of C-Type Lectin Receptor of Pattern Recognition Receptor of Immature DCs in CRC
5.2.1. Mincle
5.2.2. Dectin-3
5.2.3. DC-SIGN
5.2.4. LOX-1
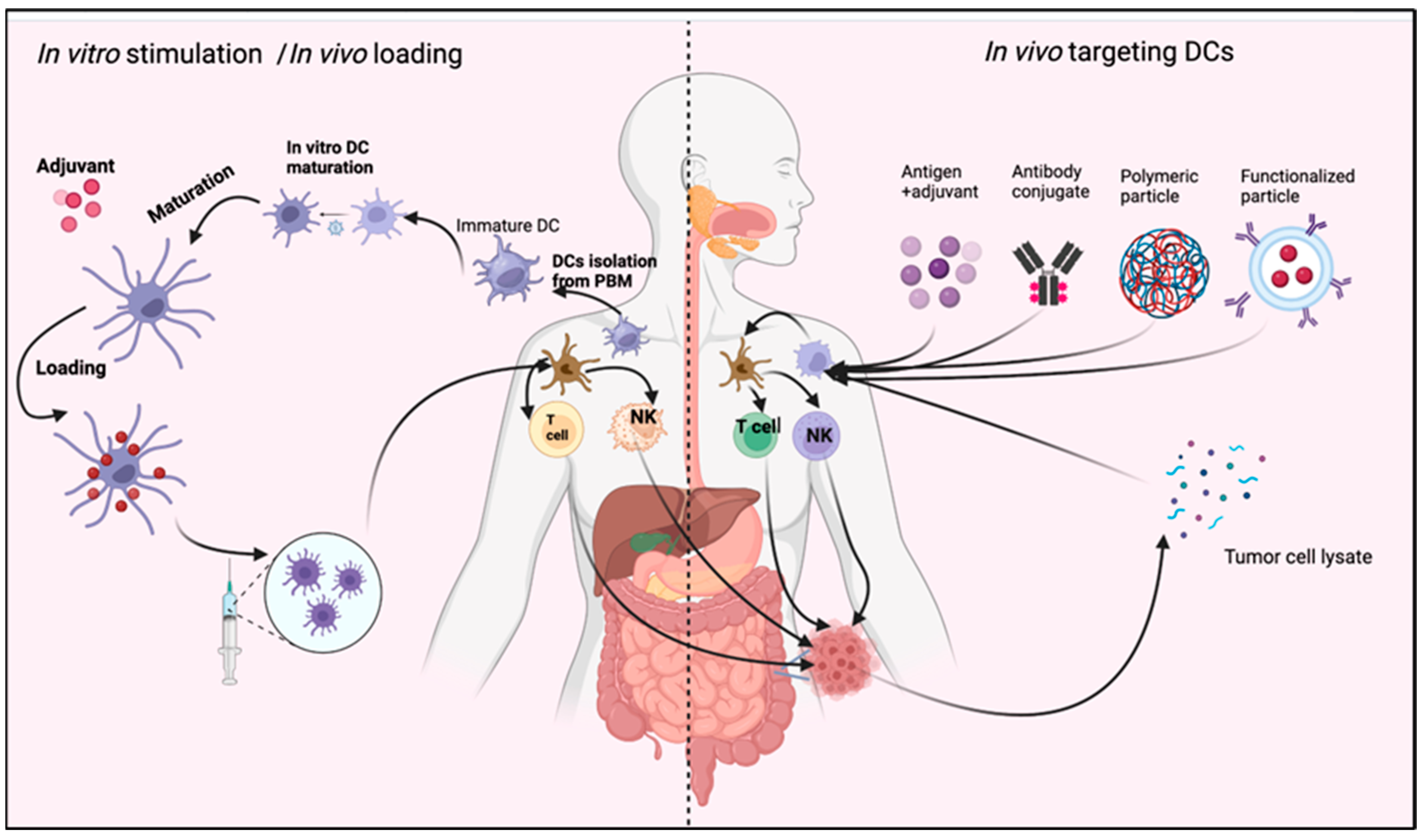
6. DCs Utilization in CRC Immunotherapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, A.K.; Divya, T.; Kumar, K.; Dineshbabu, V.; Velavan, B.; Sudhandiran, G. Colorectal carcinogenesis: Insights into the cell death and signal transduction pathways: A review. World J. Gastrointest. Oncol. 2018, 10, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Kong, C.; Liang, L.; Liu, G.; Du, L.; Yang, Y.; Liu, J.; Shi, D.; Li, X.; Ma, Y. Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer. Gut 2023, 72, 1129–1142. [Google Scholar] [CrossRef]
- Kosumi, K.; Mima, K.; Baba, H.; Ogino, S. Dysbiosis of the gut microbiota and colorectal cancer: The key target of molecular pathological epidemiology. J. Lab. Precis. Med. 2018, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Gloria, L.D.; Nannini, G.; Meoni, G.; Niccolai, E.; Ringressi, M.N.; Baldi, S.; Fani, R.; Tenori, L.; Taddei, A.; et al. From adenoma to CRC stages: The oral-gut microbiome axis as a source of potential microbial and metabolic biomarkers of malignancy. Neoplasia 2023, 40, 100901. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Z.; Zheng, W.; Liu, X.; Sun, C.; Laugsand, J.B.; Liu, Z.; Cui, G. Changes of immunocytic phenotypes and functions from human colorectal adenomatous stage to cancerous stage: Update. Immunobiology 2015, 220, 1186–1196. [Google Scholar] [CrossRef]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Wang, N.; Fang, J.Y. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2023, 31, 159–172. [Google Scholar] [CrossRef]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T. Environmental Factors, Gut Microbiota, and Colorectal Cancer Prevention. Clin. Gastroenterol. Hepatol. 2019, 17, 275–289. [Google Scholar] [CrossRef]
- American Cancer Society, CRC Facts and Figures 2020–2022. 2020. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2020-2022.pdf (accessed on 20 December 2022).
- Mariotto, A.B.; Yabroff, K.R.; Shao, Y.; Feuer, E.J.; Brown, M.L. Projections of the cost of cancer care in the United States: 2010–2020. J. Natl. Cancer Inst. 2011, 103, 117–128. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Burnett-Hartman, A.N.; Lee, J.K.; Demb, J.; Gupta, S. An Update on the Epidemiology, Molecular Characterization, Diagnosis, and Screening Strategies for Early-Onset Colorectal Cancer. Gastroenterology 2021, 160, 1041–1049. [Google Scholar] [CrossRef]
- American Institute for Cancer Research, World Cancer Research Fund, Diet, Nutrition, Physical Activity, and Colorectal Cancer. Continuous Update Projects (CUP). 2017. Available online: https://www.wcrf.org/wp-content/uploads/2021/02/Colorectal-cancer-report.pdf (accessed on 18 June 2018).
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260.e1216. [Google Scholar] [CrossRef]
- Alrahawy, M.; Javed, S.; Atif, H.; Elsanhoury, K.; Mekhaeil, K.; Eskandar, G. Microbiome and Colorectal Cancer Management. Cureus 2022, 14, e30720. [Google Scholar] [CrossRef]
- Bae, S.; Ulrich, C.M.; Neuhouser, M.L.; Malysheva, O.; Bailey, L.B.; Xiao, L.; Brown, E.C.; Cushing-Haugen, K.L.; Zheng, Y.; Cheng, T.Y.; et al. Plasma choline metabolites and colorectal cancer risk in the Women’s Health Initiative Observational Study. Cancer Res. 2014, 74, 7442–7452. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 2009, 15, 3329–3340. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132. [Google Scholar] [CrossRef]
- Ocvirk, S.; O’Keefe, S.J.D. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 2021, 73, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Norat, T.; Ferrari, P.; Jenab, M.; Bueno-de-Mesquita, B.; Skeie, G.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjønneland, A.; et al. Dietary fibre intake and risks of cancers of the colon and rectum in the European prospective investigation into cancer and nutrition (EPIC). PLoS ONE 2012, 7, e39361. [Google Scholar] [CrossRef]
- Liu, P.H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.H.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of Obesity With Risk of Early-Onset Colorectal Cancer Among Women. JAMA Oncol. 2019, 5, 37–44. [Google Scholar] [CrossRef]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef]
- Yang, B.; Bostick, R.M.; Tran, H.Q.; Gewirtz, A.T.; Campbell, P.T.; Fedirko, V. Circulating Biomarkers of Gut Barrier Function: Correlates and Nonresponse to Calcium Supplementation among Colon Adenoma Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Luo, F.K.; Wang, Y.L.; Tang, J.L.; Liu, Y.S. LBP and CD14 polymorphisms correlate with increased colorectal carcinoma risk in Han Chinese. World J. Gastroenterol. 2011, 17, 2326–2331. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med. Sci. Sports Exerc. 2018, 50, 747–757. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.V.; de Camargo, M.R.; Russo, E.; Amedei, A. Role of diet and gut microbiota on colorectal cancer immunomodulation. World J. Gastroenterol. 2019, 25, 151–162. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huipeng, W.; Lifeng, G.; Chuang, G.; Jiaying, Z.; Yuankun, C. The differences in colonic mucosal microbiota between normal individual and colon cancer patients by polymerase chain reaction-denaturing gradient gel electrophoresis. J. Clin. Gastroenterol. 2014, 48, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum-symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv. Protein Chem. Struct. Biol. 2020, 120, 45–84. [Google Scholar] [CrossRef] [PubMed]
- Rossi, O.; van Baarlen, P.; Wells, J.M. Host-recognition of pathogens and commensals in the mammalian intestine. Curr. Top. Microbiol. Immunol. 2013, 358, 291–321. [Google Scholar] [CrossRef]
- Mantri, C.K.; Chen, C.H.; Dong, X.; Goodwin, J.S.; Pratap, S.; Paromov, V.; Xie, H. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 2015, 4, 53–65. [Google Scholar] [CrossRef]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29. [Google Scholar] [CrossRef]
- Wilensky, A.; Tzach-Nahman, R.; Potempa, J.; Shapira, L.; Nussbaum, G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J. Innate Immun. 2015, 7, 127–135. [Google Scholar] [CrossRef]
- Gaddis, D.E.; Maynard, C.L.; Weaver, C.T.; Michalek, S.M.; Katz, J. Role of TLR2-dependent IL-10 production in the inhibition of the initial IFN-γ T cell response to Porphyromonas gingivalis. J. Leukoc. Biol. 2013, 93, 21–31. [Google Scholar] [CrossRef]
- Cerboni, S.; Gentili, M.; Manel, N. Diversity of pathogen sensors in dendritic cells. Adv. Immunol. 2013, 120, 211–237. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The control of immunity and tolerance by dendritic cell. Pathol. Biol. 2003, 51, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Luft, T.; Rodionova, E.; Maraskovsky, E.; Kirsch, M.; Hess, M.; Buchholtz, C.; Goerner, M.; Schnurr, M.; Skoda, R.; Ho, A.D. Adaptive functional differentiation of dendritic cells: Integrating the network of extra- and intracellular signals. Blood 2006, 107, 4763–4769. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N. Dendritic cells: A conductor of T cell differentiation. Allergol. Int. 2007, 56, 193–199. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Perona-Wright, G.; Worsley, A.G.; Ishii, N.; MacDonald, A.S. Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J. Immunol. 2007, 179, 3515–3523. [Google Scholar] [CrossRef]
- Huang, G.; Wang, Y.; Chi, H. Regulation of TH17 cell differentiation by innate immune signals. Cell Mol. Immunol. 2012, 9, 287–295. [Google Scholar] [CrossRef]
- Levings, M.K.; Gregori, S.; Tresoldi, E.; Cazzaniga, S.; Bonini, C.; Roncarolo, M.G. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005, 105, 1162–1169. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Pérez-Gracia, J.L.; Sánchez-Arráez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28 (Suppl. S12), xii74. [Google Scholar] [CrossRef]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef]
- Rock, K.L.; Shen, L. Cross-presentation: Underlying mechanisms and role in immune surveillance. Immunol. Rev. 2005, 207, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Valladeau, J.; Ravel, O.; Dezutter-Dambuyant, C.; Moore, K.; Kleijmeer, M.; Liu, Y.; Duvert-Frances, V.; Vincent, C.; Schmitt, D.; Davoust, J.; et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000, 12, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’Amico, G.; Stoppacciaro, A.; Mancinelli, R.; van’t Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: Selective expression of TLR3 in dendritic cells. J. Immunol. 2000, 164, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Granucci, F.; Ricciardi-Castagnoli, P. Molecular events of bacterial-induced maturation of dendritic cells. J. Clin. Immunol. 2000, 20, 161–166. [Google Scholar] [CrossRef]
- Wilson, N.S.; El-Sukkari, D.; Villadangos, J.A. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood 2004, 103, 2187–2195. [Google Scholar] [CrossRef]
- Deluce-Kakwata-Nkor, N.; Lamendour, L.; Chabot, V.; Héraud, A.; Ivanovic, Z.; Halary, F.; Dehaut, F.; Velge-Roussel, F. Differentiation of human dendritic cell subsets for immune tolerance induction. Transfus. Clin. Biol. 2018, 25, 90–95. [Google Scholar] [CrossRef]
- Wallet, M.A.; Sen, P.; Flores, R.R.; Wang, Y.; Yi, Z.; Huang, Y.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; et al. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008, 205, 219–232. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Kaplan, C.W.; Ma, X.; Paranjpe, A.; Jewett, A.; Lux, R.; Kinder-Haake, S.; Shi, W. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 2010, 78, 4773–4778. [Google Scholar] [CrossRef]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H.; et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef]
- Spigaglia, P.; Barbanti, F.; Germinario, E.A.P.; Criscuolo, E.M.; Bruno, G.; Sanchez-Mete, L.; Porowska, B.; Stigliano, V.; Accarpio, F.; Oddi, A.; et al. Comparison of microbiological profile of enterotoxigenic Bacteroides fragilis (ETBF) isolates from subjects with colorectal cancer (CRC) or intestinal pre-cancerous lesions versus healthy individuals and evaluation of environmental factors involved in intestinal dysbiosis. Anaerobe 2023, 82, 102757. [Google Scholar] [CrossRef]
- Dejea, C.; Wick, E.; Sears, C.L. Bacterial oncogenesis in the colon. Future Microbiol. 2013, 8, 445–460. [Google Scholar] [CrossRef]
- Ren, L.; Ye, J.; Zhao, B.; Sun, J.; Cao, P.; Yang, Y. The Role of Intestinal Microbiota in Colorectal Cancer. Front. Pharmacol. 2021, 12, 674807. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Colorectal Cancer Early Detection, Diagnosis, and Staging. cancer.org | 1.800.227.2345. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/8606.00.pdf (accessed on 3 June 2022).
- Lochhead, P.; Chan, A.T.; Giovannucci, E.; Fuchs, C.S.; Wu, K.; Nishihara, R.; O’Brien, M.; Ogino, S. Progress and opportunities in molecular pathological epidemiology of colorectal premalignant lesions. Am. J. Gastroenterol. 2014, 109, 1205–1214. [Google Scholar] [CrossRef]
- Frank, S.A. Incidence, Inheritance, and Evolution. In Dynamics of Cancer; Princeton University Press: Princeton, NJ, USA, 2007. [Google Scholar]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Bateman, A.C. Pathology of serrated colorectal lesions. J. Clin. Pathol. 2014, 67, 865–874. [Google Scholar] [CrossRef]
- Kang, M.; Edmundson, P.; Araujo-Perez, F.; McCoy, A.N.; Galanko, J.; Keku, T.O. Association of plasma endotoxin, inflammatory cytokines and risk of colorectal adenomas. BMC Cancer 2013, 13, 91. [Google Scholar] [CrossRef]
- Melotte, V.; Lentjes, M.H.; van den Bosch, S.M.; Hellebrekers, D.M.; de Hoon, J.P.; Wouters, K.A.; Daenen, K.L.; Partouns-Hendriks, I.E.; Stessels, F.; Louwagie, J.; et al. N-Myc downstream-regulated gene 4 (NDRG4): A candidate tumor suppressor gene and potential biomarker for colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 916–927. [Google Scholar] [CrossRef]
- Yamane, L.; Scapulatempo-Neto, C.; Reis, R.M.; Guimarães, D.P. Serrated pathway in colorectal carcinogenesis. World J. Gastroenterol. 2014, 20, 2634–2640. [Google Scholar] [CrossRef]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar] [CrossRef]
- Solano-Gálvez, S.G.; Tovar-Torres, S.M.; Tron-Gómez, M.S.; Weiser-Smeke, A.E.; Álvarez-Hernández, D.A.; Franyuti-Kelly, G.A.; Tapia-Moreno, M.; Ibarra, A.; Gutiérrez-Kobeh, L.; Vázquez-López, R. Human Dendritic Cells: Ontogeny and Their Subsets in Health and Disease. Med. Sci. 2018, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Legitimo, A.; Consolini, R.; Failli, A.; Orsini, G.; Spisni, R. Dendritic cell defects in the colorectal cancer. Hum. Vaccin. Immunother. 2014, 10, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef]
- Wu, C.; Shao, Y.; Gu, W. Immunotherapy combined with radiotherapy to reverse immunosuppression in microsatellite stable colorectal cancer. Clin. Transl. Oncol. 2023, 25, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Del Cornò, M.; D’Archivio, M.; Conti, L.; Scazzocchio, B.; Varì, R.; Donninelli, G.; Varano, B.; Giammarioli, S.; De Meo, S.; Silecchia, G.; et al. Visceral fat adipocytes from obese and colorectal cancer subjects exhibit distinct secretory and ω6 polyunsaturated fatty acid profiles and deliver immunosuppressive signals to innate immunity cells. Oncotarget 2016, 7, 63093–63105. [Google Scholar] [CrossRef] [PubMed]
- Donninelli, G.; Del Cornò, M.; Pierdominici, M.; Scazzocchio, B.; Varì, R.; Varano, B.; Pacella, I.; Piconese, S.; Barnaba, V.; D’Archivio, M.; et al. Distinct Blood and Visceral Adipose Tissue Regulatory T Cell and Innate Lymphocyte Profiles Characterize Obesity and Colorectal Cancer. Front. Immunol. 2017, 8, 643. [Google Scholar] [CrossRef]
- Gulubova, M.V.; Ananiev, J.R.; Vlaykova, T.I.; Yovchev, Y.; Tsoneva, V.; Manolova, I.M. Role of dendritic cells in progression and clinical outcome of colon cancer. Int. J. Color. Dis. 2012, 27, 159–169. [Google Scholar] [CrossRef]
- Nagorsen, D.; Voigt, S.; Berg, E.; Stein, H.; Thiel, E.; Loddenkemper, C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: Relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J. Transl. Med. 2007, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, T.; Weiss, J.E.; Schned, A.R.; Barth, R.J., Jr. Dendritic cell infiltration in colon cancer. J. Immunother. 2001, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Michielsen, A.J.; Noonan, S.; Martin, P.; Tosetto, M.; Marry, J.; Biniecka, M.; Maguire, A.A.; Hyland, J.M.; Sheahan, K.D.; O’Donoghue, D.P.; et al. Inhibition of dendritic cell maturation by the tumor microenvironment correlates with the survival of colorectal cancer patients following bevacizumab treatment. Mol. Cancer Ther. 2012, 11, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.; Michel, S.; Reuschenbach, M.; Nelius, N.; von Knebel Doeberitz, M.; Kloor, M. Dendritic cell and macrophage infiltration in microsatellite-unstable and microsatellite-stable colorectal cancer. Fam. Cancer 2011, 10, 557–565. [Google Scholar] [CrossRef]
- Gutting, T.; Burgermeister, E.; Härtel, N.; Ebert, M.P. Checkpoints and beyond-Immunotherapy in colorectal cancer. Semin. Cancer Biol. 2019, 55, 78–89. [Google Scholar] [CrossRef]
- Orsini, G.; Legitimo, A.; Failli, A.; Ferrari, P.; Nicolini, A.; Spisni, R.; Miccoli, P.; Consolini, R. Defective generation and maturation of dendritic cells from monocytes in colorectal cancer patients during the course of disease. Int. J. Mol. Sci. 2013, 14, 22022–22041. [Google Scholar] [CrossRef]
- Della Porta, M.; Danova, M.; Rigolin, G.M.; Brugnatelli, S.; Rovati, B.; Tronconi, C.; Fraulini, C.; Russo Rossi, A.; Riccardi, A.; Castoldi, G. Dendritic cells and vascular endothelial growth factor in colorectal cancer: Correlations with clinicobiological findings. Oncology 2005, 68, 276–284. [Google Scholar] [CrossRef]
- Kvistborg, P.; Bechmann, C.M.; Pedersen, A.W.; Toh, H.C.; Claesson, M.H.; Zocca, M.B. Comparison of monocyte-derived dendritic cells from colorectal cancer patients, non-small-cell-lung-cancer patients and healthy donors. Vaccine 2009, 28, 542–547. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Amicarella, F.; Muraro, M.G.; Hirt, C.; Cremonesi, E.; Padovan, E.; Mele, V.; Governa, V.; Han, J.; Huber, X.; Droeser, R.A.; et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut 2017, 66, 692–704. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Yuan, A.; Goll, R.; Florholmen, J. IL-17A in the tumor microenvironment of the human colorectal adenoma-carcinoma sequence. Scand. J. Gastroenterol. 2012, 47, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Monin, L.; Gaffen, S.L. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef] [PubMed]
- Li, T.H.; Liu, L.; Hou, Y.Y.; Shen, S.N.; Wang, T.T. C-type lectin receptor-mediated immune recognition and response of the microbiota in the gut. Gastroenterol. Rep. 2019, 7, 312–321. [Google Scholar] [CrossRef]
- Navarro-Arias, M.J.; Hernández-Chávez, M.J.; García-Carnero, L.C.; Amezcua-Hernández, D.G.; Lozoya-Pérez, N.E.; Estrada-Mata, E.; Martínez-Duncker, I.; Franco, B.; Mora-Montes, H.M. Differential recognition of Candida tropicalis, Candida guilliermondii, Candida krusei, and Candida auris by human innate immune cells. Infect. Drug Resist. 2019, 12, 783–794. [Google Scholar] [CrossRef]
- Del Fresno, C.; Iborra, S.; Saz-Leal, P.; Martínez-López, M.; Sancho, D. Flexible Signaling of Myeloid C-Type Lectin Receptors in Immunity and Inflammation. Front. Immunol. 2018, 9, 804. [Google Scholar] [CrossRef]
- Pan, Y.G.; Yu, Y.L.; Lin, C.C.; Lanier, L.L.; Chu, C.L. FcεRI γ-Chain Negatively Modulates Dectin-1 Responses in Dendritic Cells. Front. Immunol. 2017, 8, 1424. [Google Scholar] [CrossRef]
- Martínez-López, M.; Iborra, S.; Conde-Garrosa, R.; Mastrangelo, A.; Danne, C.; Mann, E.R.; Reid, D.M.; Gaboriau-Routhiau, V.; Chaparro, M.; Lorenzo, M.P.; et al. Microbiota Sensing by Mincle-Syk Axis in Dendritic Cells Regulates Interleukin-17 and -22 Production and Promotes Intestinal Barrier Integrity. Immunity 2019, 50, 446–461.e9. [Google Scholar] [CrossRef]
- Leonardi, I.; Li, X.; Semon, A.; Li, D.; Doron, I.; Putzel, G.; Bar, A.; Prieto, D.; Rescigno, M.; McGovern, D.P.B.; et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 2018, 359, 232–236. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, T.; Lu, X.; Xu, Z.; Qu, J.; Zhang, Z.; Shi, G.; Shen, S.; Hou, Y.; Chen, Y.; et al. Fungal-induced glycolysis in macrophages promotes colon cancer by enhancing innate lymphoid cell secretion of IL-22. Embo J. 2021, 40, e105320. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, X.; Zhang, J.; Wang, K.; Zhang, Y.; Shang, W.; Zhang, Y.; Cui, J.; Shi, X.; Na, H.; et al. DC-SIGN-LEF1/TCF1-miR-185 feedback loop promotes colorectal cancer invasion and metastasis. Cell Death Differ. 2020, 27, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Fernandes, Â.; Oliveira, M.; Resende, C.; Correia, A.; de-Freitas-Junior, J.C.; Lavelle, A.; Andrade-da-Costa, J.; Leander, M.; Xavier-Ferreira, H.; et al. Glycans as Immune Checkpoints: Removal of Branched N-glycans Enhances Immune Recognition Preventing Cancer Progression. Cancer Immunol. Res. 2020, 8, 1407–1425. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, R.; Li, J.; Li, J. The Role of C-Type Lectin Receptor Signaling in the Intestinal Microbiota-Inflammation-Cancer Axis. Front. Immunol. 2022, 13, 894445. [Google Scholar] [CrossRef]
- Wang, C.; Barnoud, C.; Cenerenti, M.; Sun, M.; Caffa, I.; Kizil, B.; Bill, R.; Liu, Y.; Pick, R.; Garnier, L.; et al. Dendritic cells direct circadian anti-tumour immune responses. Nature 2023, 614, 136–143. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef] [PubMed]
- Aldahlawi, A.M.; Abdullah, S.T. Dendritic Cell-Based Immunotherapies and their Potential use in Colorectal Cancer Immunotherapy. J. Microsc. Ultrastruct. 2022, 10, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Poirier, A.; Tremblay, M.L. Pharmacological potentiation of monocyte-derived dendritic cell cancer immunotherapy. Cancer Immunol. Immunother. 2023, 72, 1343–1353. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaher, K.; Basingab, F. Interaction between Gut Microbiota and Dendritic Cells in Colorectal Cancer. Biomedicines 2023, 11, 3196. https://doi.org/10.3390/biomedicines11123196
Zaher K, Basingab F. Interaction between Gut Microbiota and Dendritic Cells in Colorectal Cancer. Biomedicines. 2023; 11(12):3196. https://doi.org/10.3390/biomedicines11123196
Chicago/Turabian StyleZaher, Kawther, and Fatemah Basingab. 2023. "Interaction between Gut Microbiota and Dendritic Cells in Colorectal Cancer" Biomedicines 11, no. 12: 3196. https://doi.org/10.3390/biomedicines11123196
APA StyleZaher, K., & Basingab, F. (2023). Interaction between Gut Microbiota and Dendritic Cells in Colorectal Cancer. Biomedicines, 11(12), 3196. https://doi.org/10.3390/biomedicines11123196