
Targeting Autophagy, Apoptosis, and Oxidative Perturbations with Dapagliflozin Mitigates Cadmium-Induced Cognitive Dysfunction in Rats

 , , , ,
, , , ,
Abstract
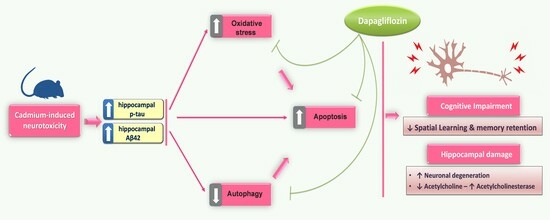
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Experimental Animals
2.3. Preclinical Animal Model
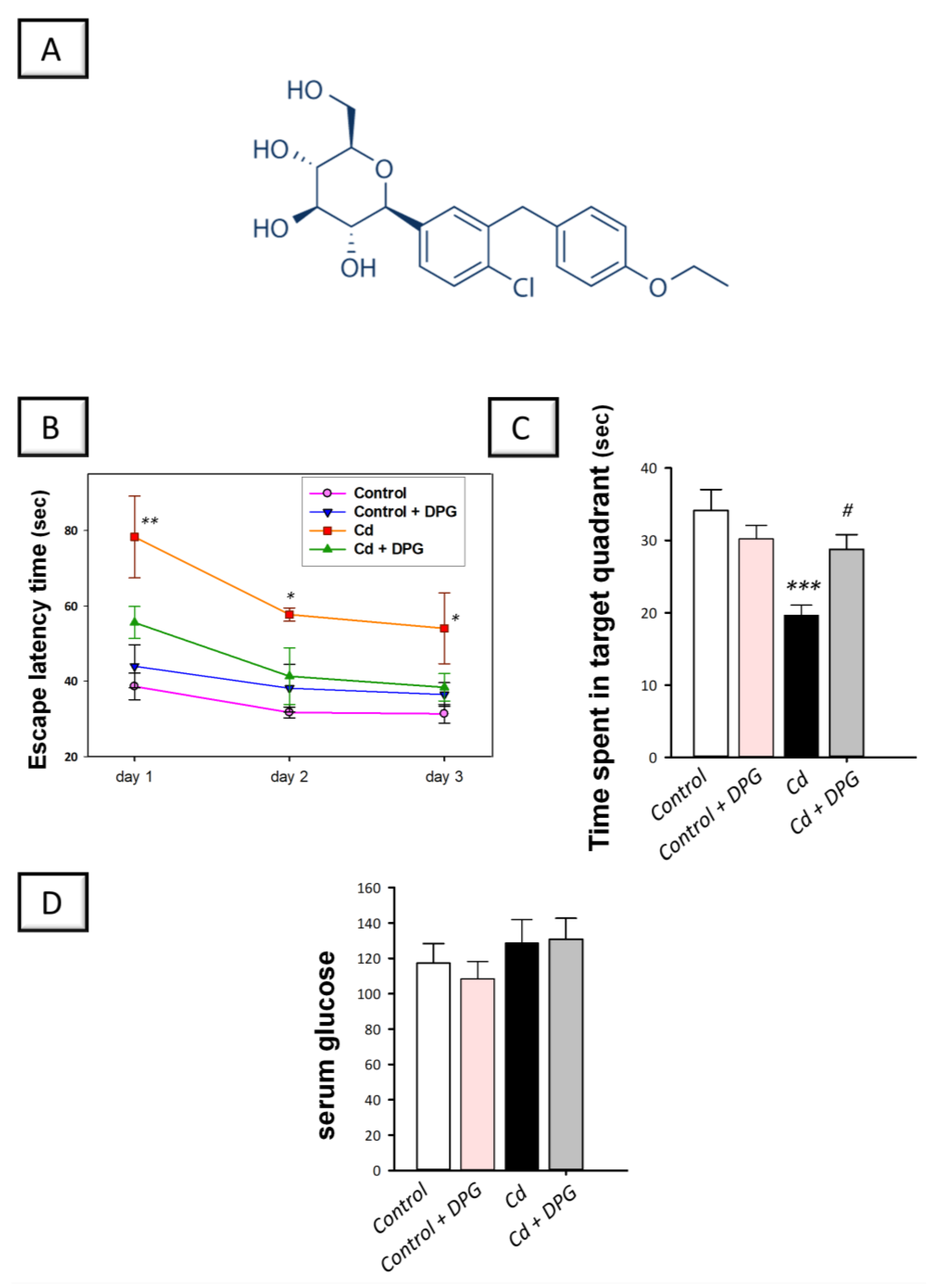
2.4. Morris Water Maze (MWM)
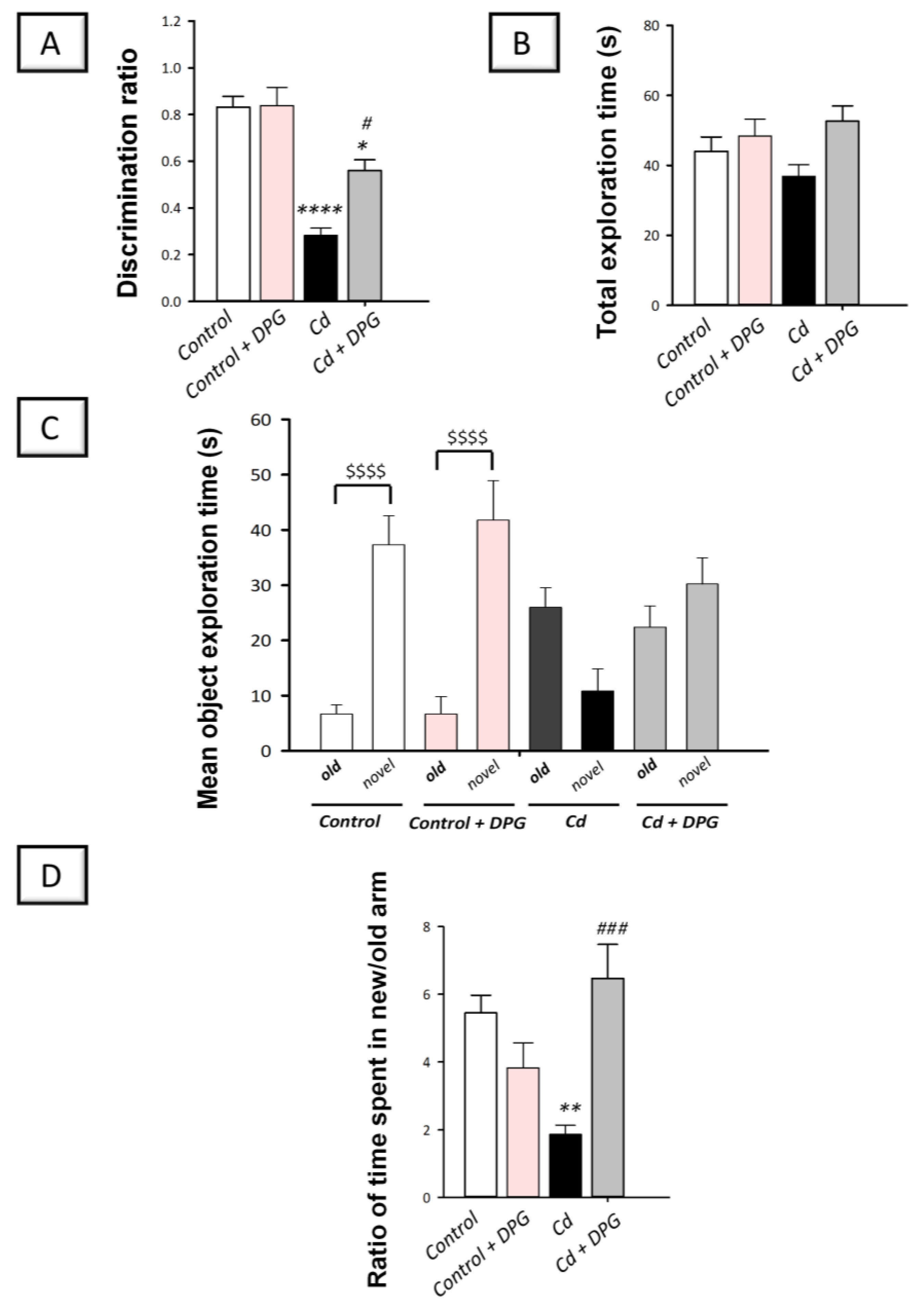
2.5. Novel Object Recognition Test (NORT)
2.6. Y-Shaped Maze
2.7. Collecting and Processing Brain Tissue
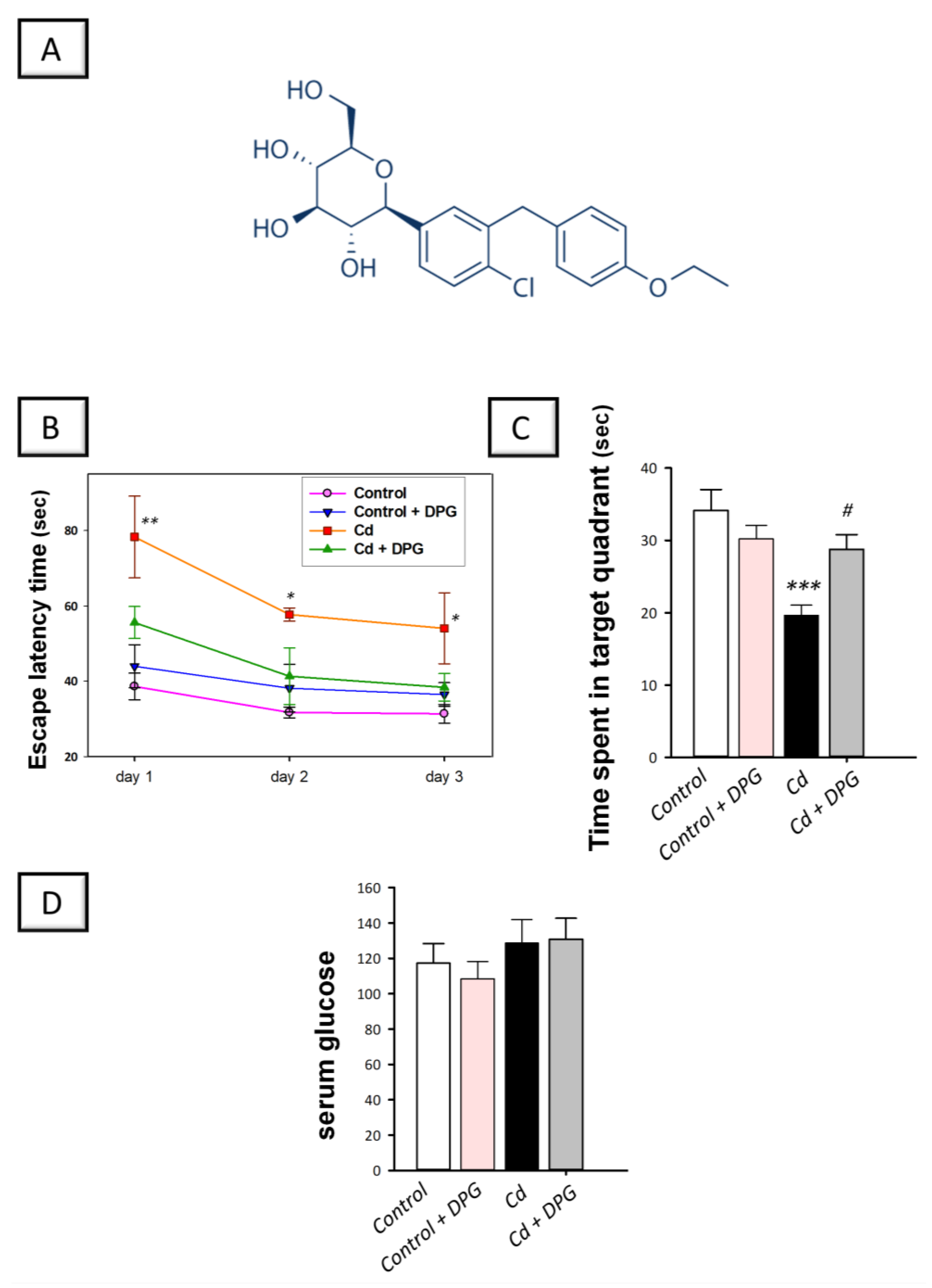
2.8. Measurement of Serum Glucose
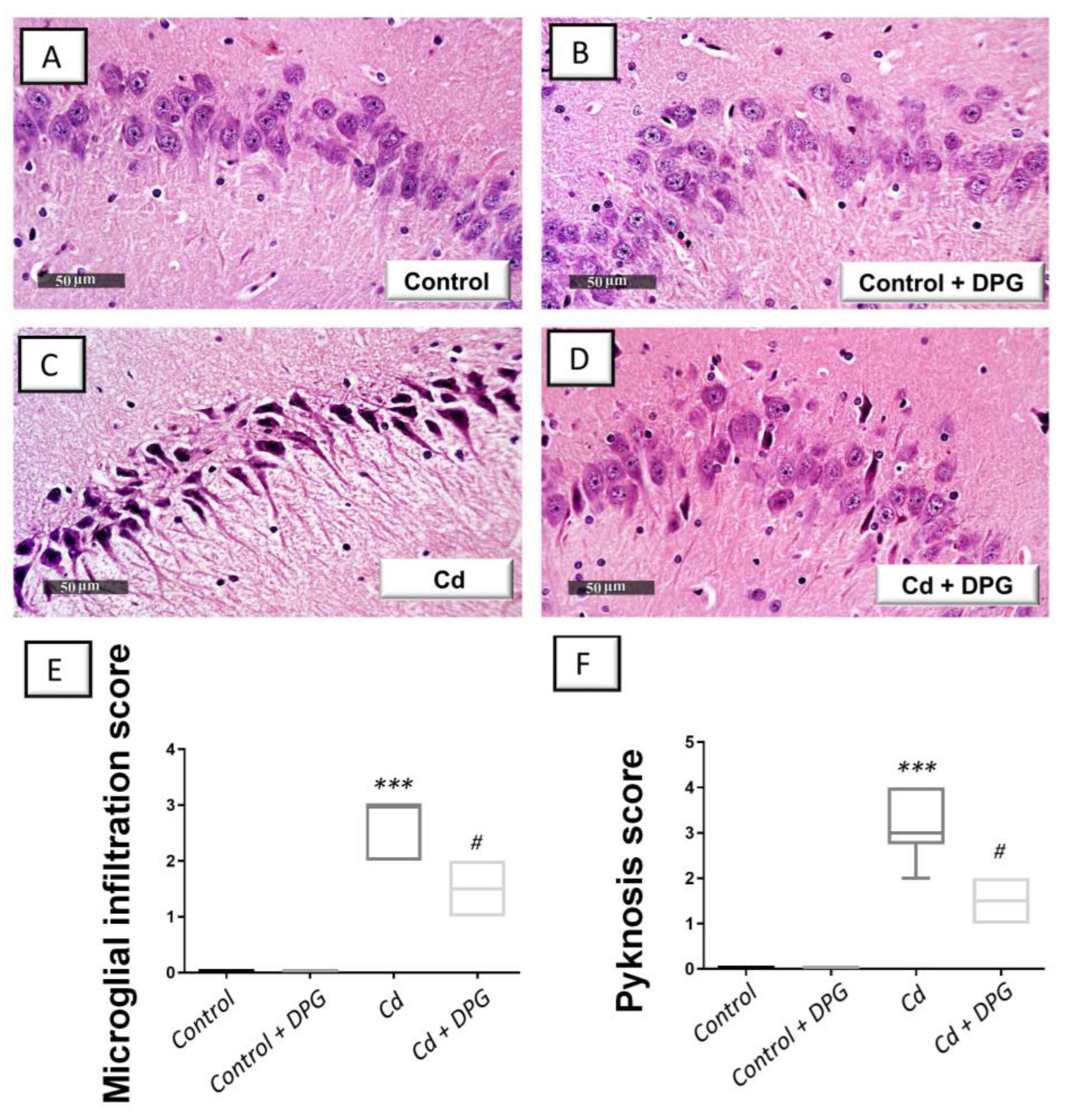
2.9. Histopathology
2.10. Immunohistochemistry
2.11. Evaluation of Acetylcholine Esterase, Acetylcholine, and Caspase-3 Activity
2.12. Evaluation of Hippocampal p-tau, Aβ42, and p-GSK-3β
2.13. Determination of Autophagy Events
2.14. Evaluation of the Redox Milieu
2.15. Data Presentation and Statistical Analysis
3. Results
3.1. Dapagliflozin Improved Memory Retention in Cadmium-Intoxicated Rats
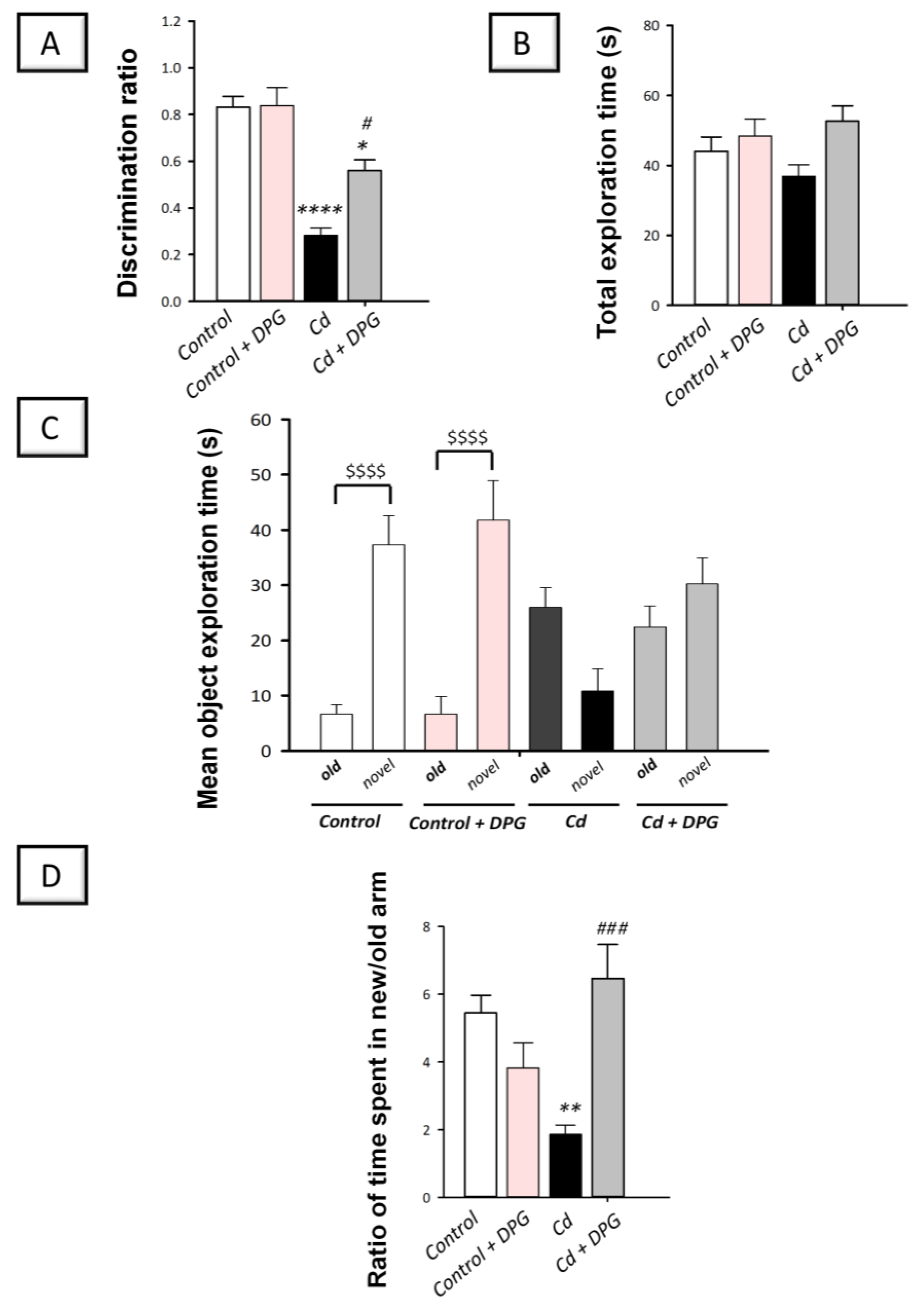
3.2. Dapagliflozin Rescued Recognition Memory Deterioration in Cadmium-Intoxicated Rats
3.3. Dapagliflozin Mitigated Hippocampal Histopathological Signs in Cadmium-Intoxicated Rats
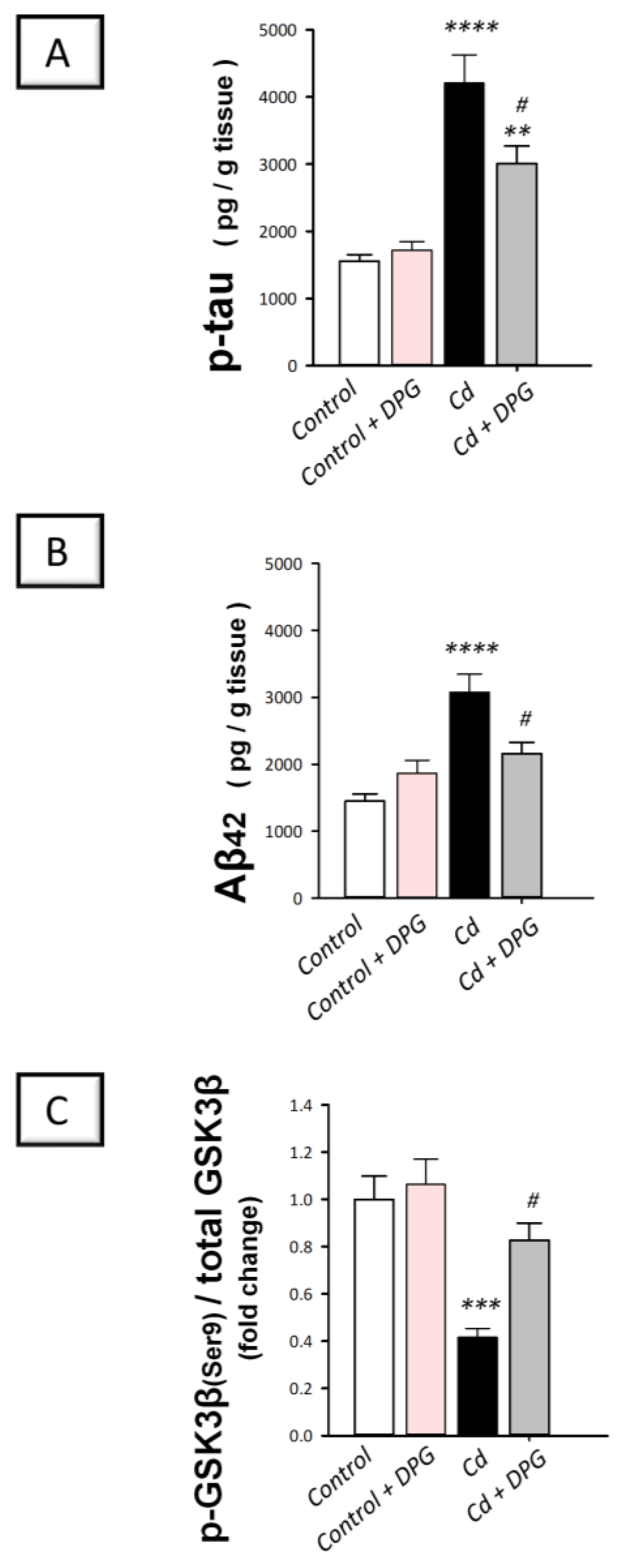
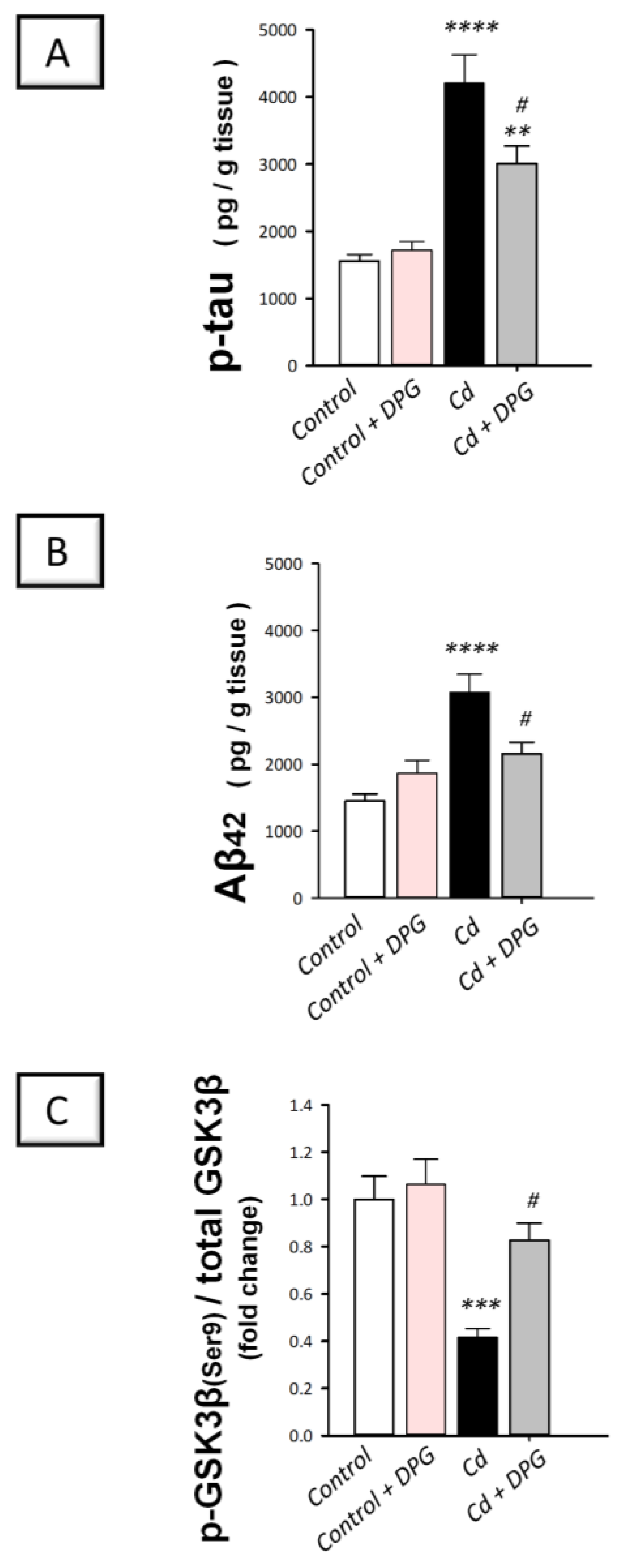
3.4. Dapagliflozin Downregulated Hippocampal Protein Expression of p-tau and Aβ42 While Upregulating the Inactive p-GSK-3β(Ser9) in Cadmium-Intoxicated Rats
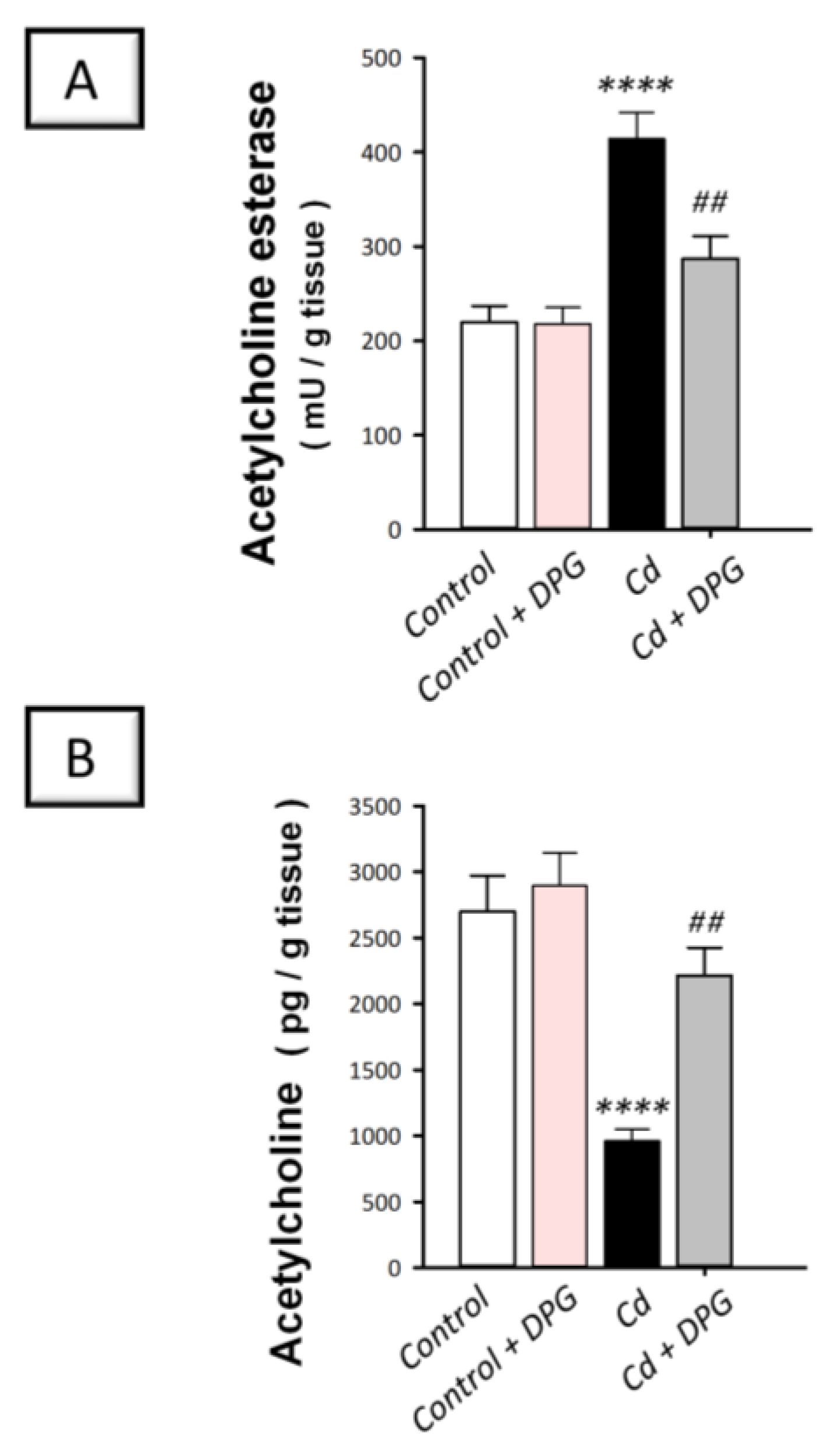
3.5. Dapagliflozin Attenuated Hippocampal Acetylcholine Esterase While Enhancing Acetylcholine Levels in Cadmium-Intoxicated Rats
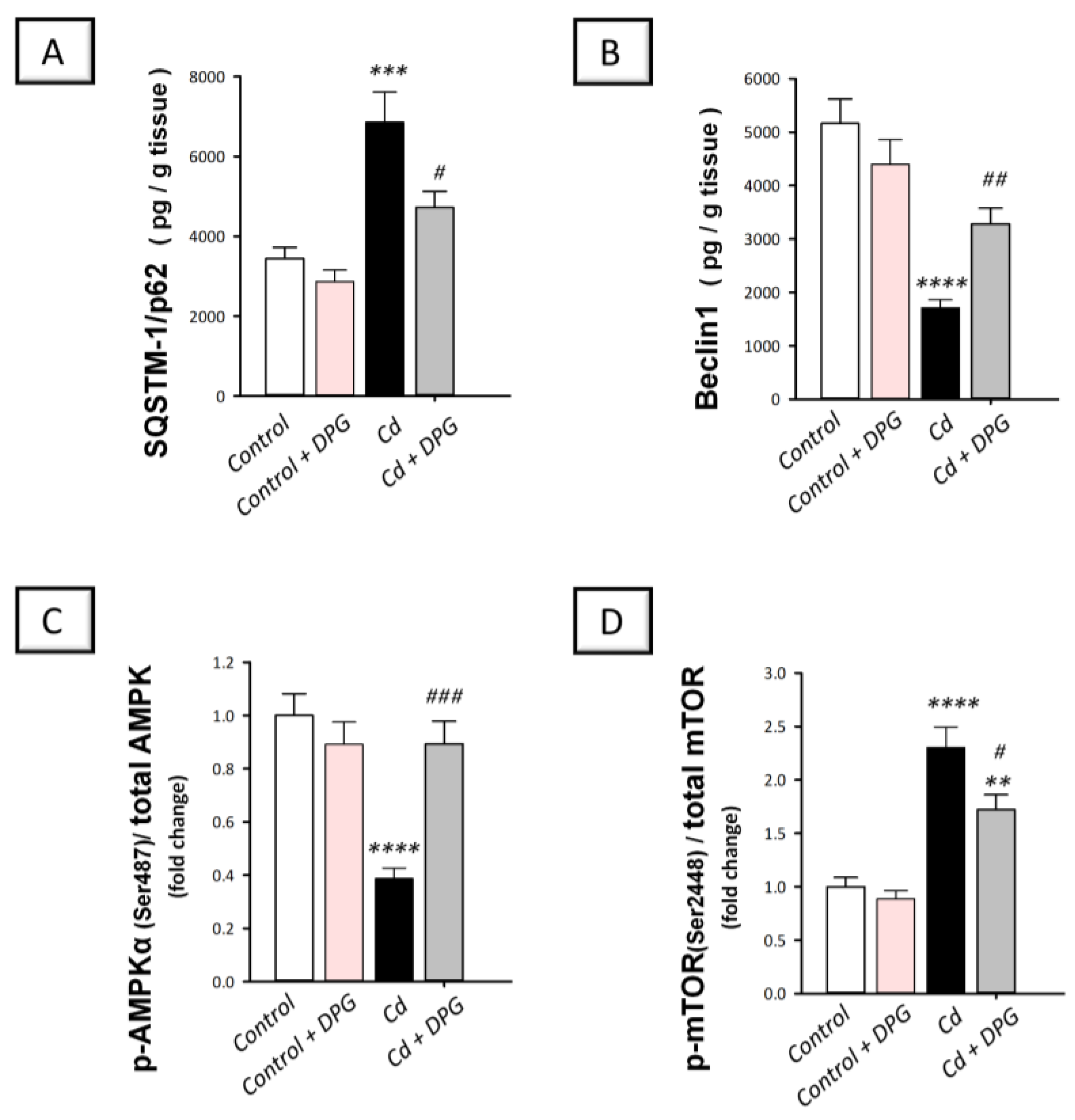
3.6. Dapagliflozin Stimulated Hippocampal Impaired Autophagy Events and Stimulated AMPK/mTOR Pathway in Cadmium-Intoxicated Rats
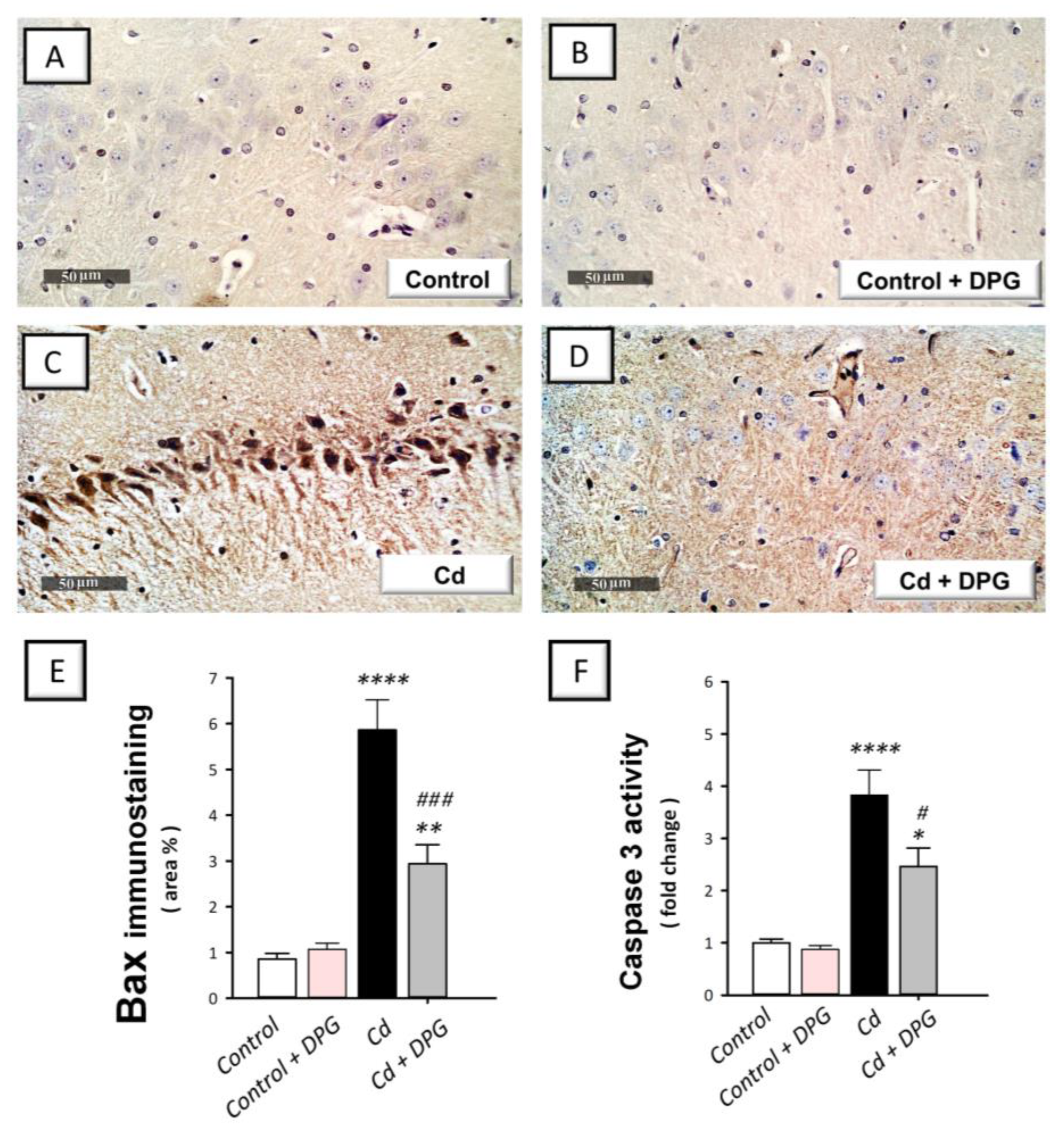
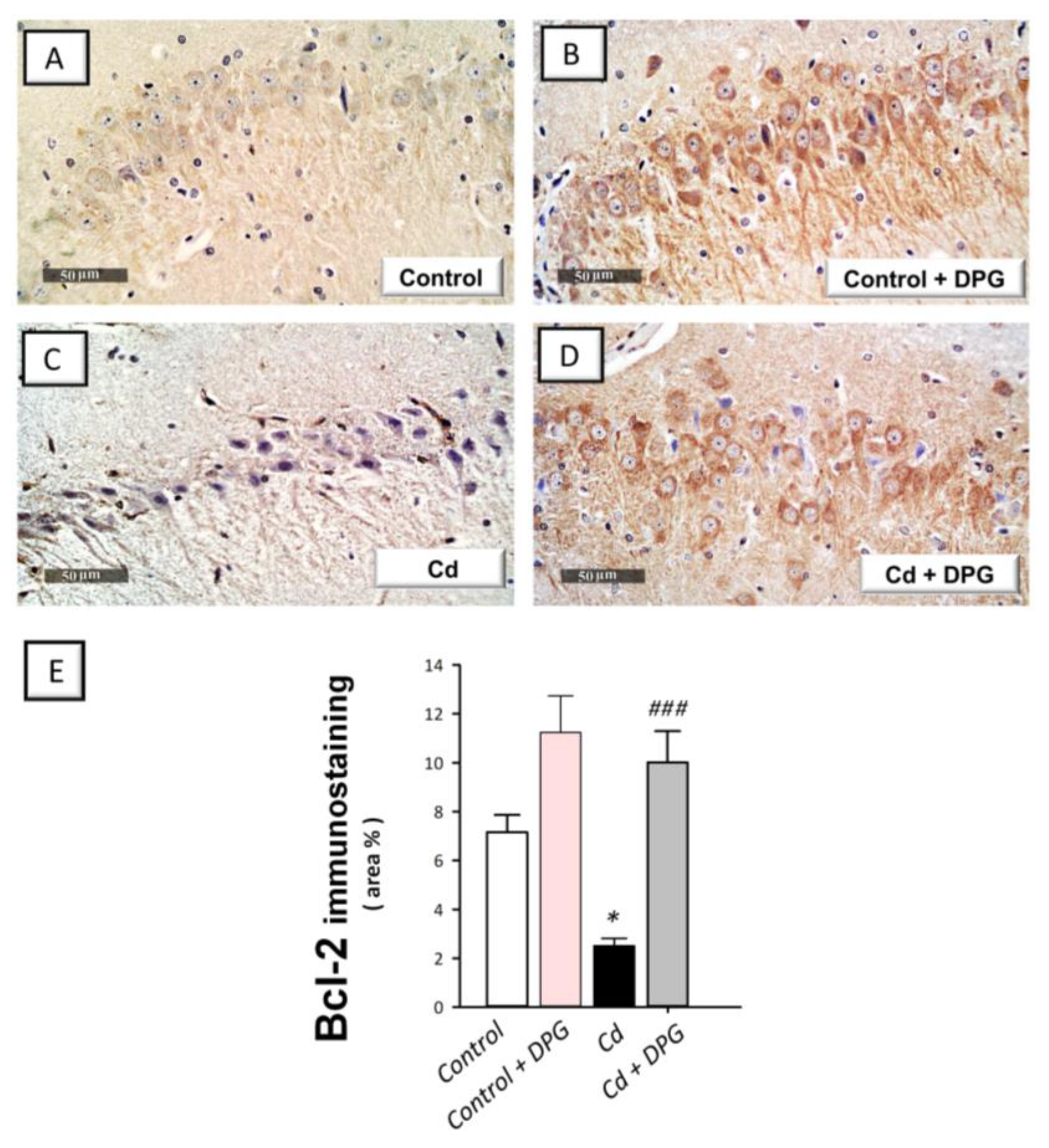
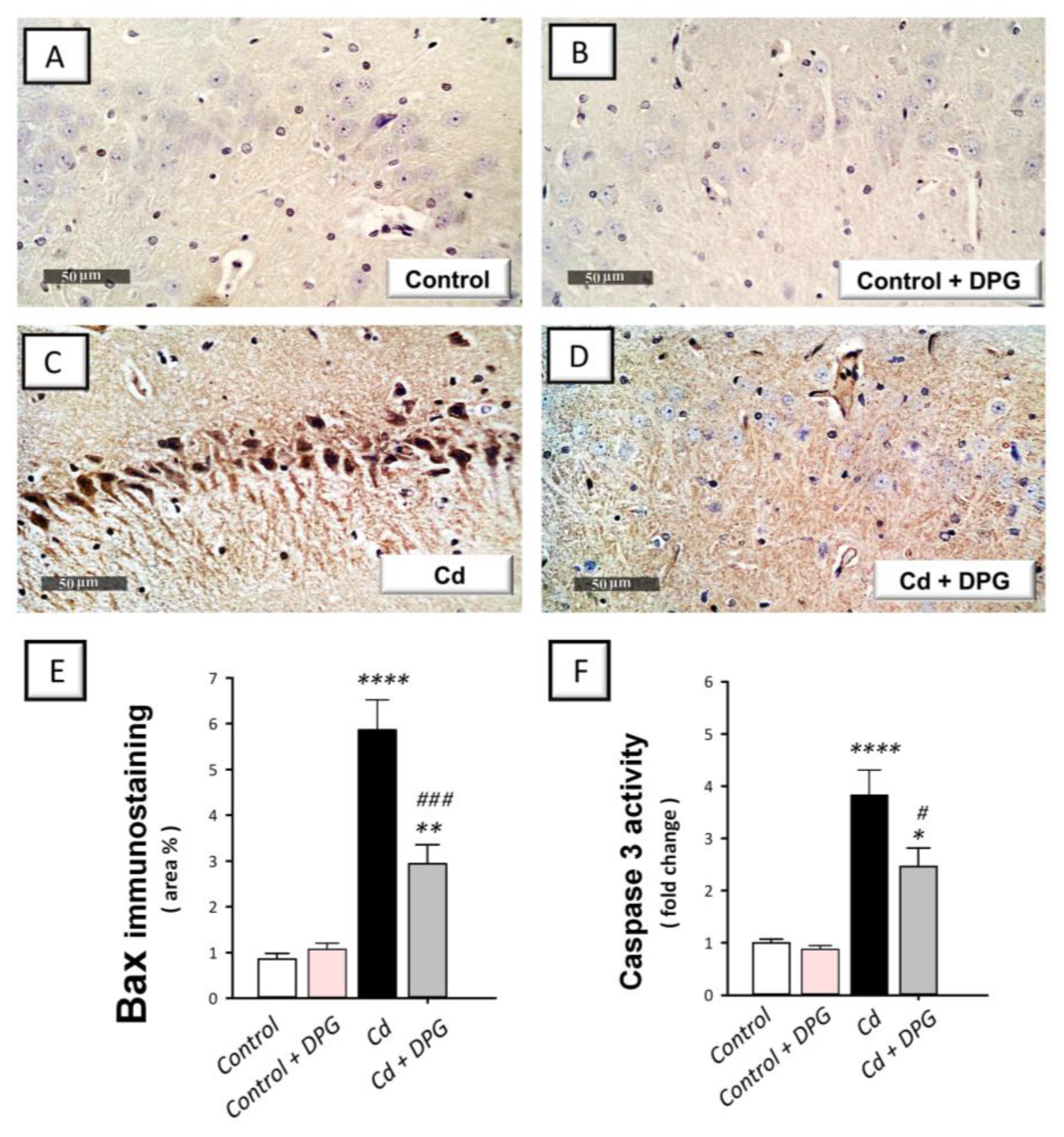
3.7. Dapagliflozin Reversed the Apoptotic Events in Cadmium-Intoxicated Rats
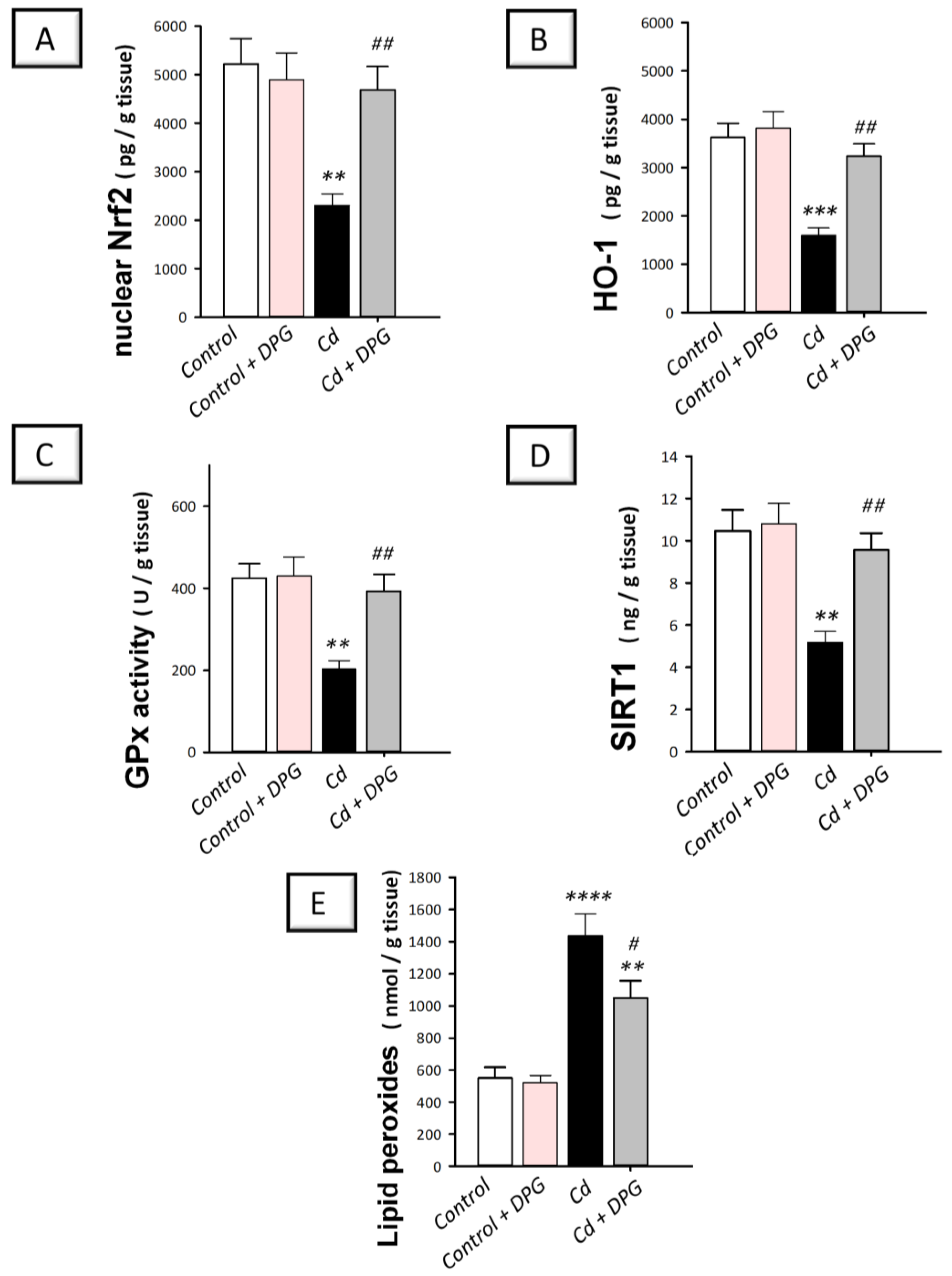
3.8. Dapagliflozin Reversed Hippocampal Pro-Oxidant Events in Cadmium-Intoxicated Rats
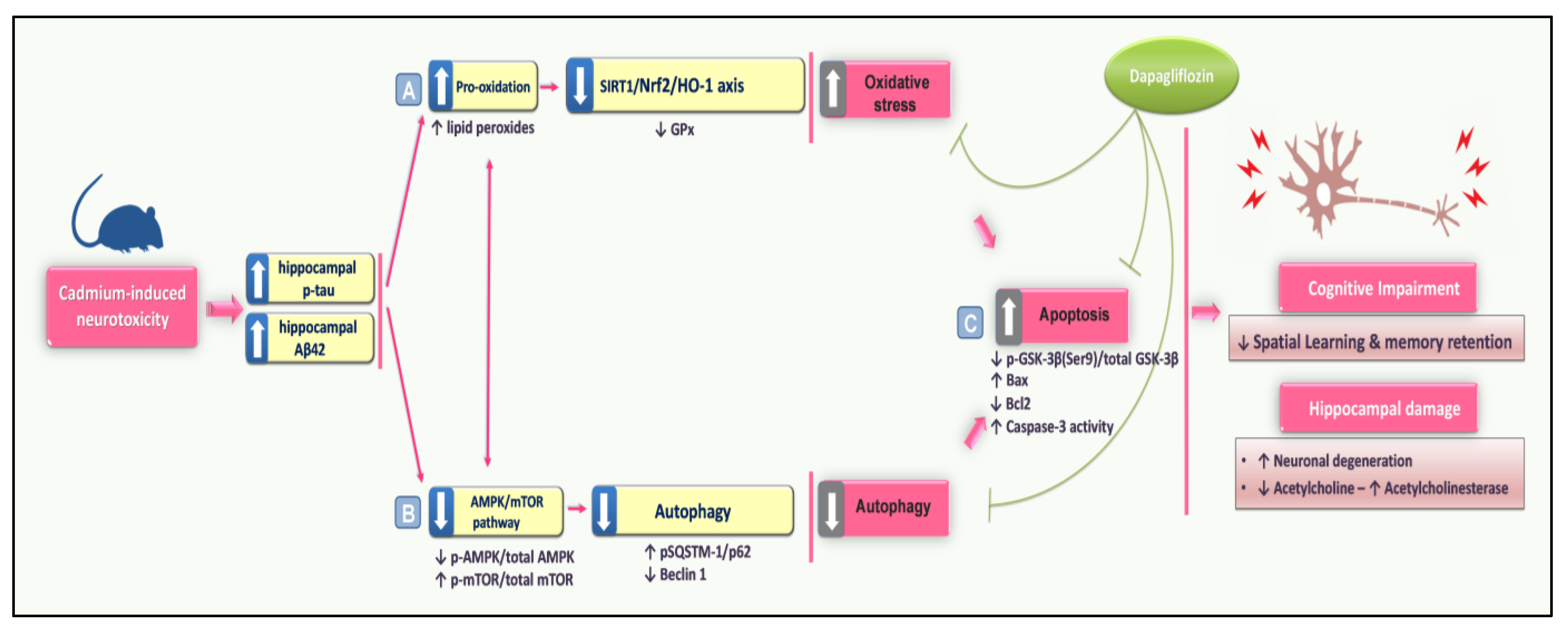
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimers Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.; Lee, H.K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef]
- Ali, T.; Khan, A.; Alam, S.I.; Ahmad, S.; Ikram, M.; Park, J.S.; Lee, H.J.; Kim, M.O. Cadmium, an Environmental Contaminant, Exacerbates Alzheimer’s Pathology in the Aged Mice’s Brain. Front. Aging Neurosci. 2021, 13, 650930. [Google Scholar] [CrossRef]
- Hao, R.; Song, X.; Li, F.; Tan, X.; Sun-Waterhouse, D.; Li, D. Caffeic acid phenethyl ester reversed cadmium-induced cell death in hippocampus and cortex and subsequent cognitive disorders in mice: Involvements of AMPK/SIRT1 pathway and amyloid-tau-neuroinflammation axis. Food Chem. Toxicol. 2020, 144, 111636. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.K.; Liu, X.Y.; Wang, Z.Y.; Qu, K.C.; Fan, R.F. Trehalose alleviates cadmium-induced brain damage by ameliorating oxidative stress, autophagy inhibition, and apoptosis. Metallomics 2019, 11, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Wu, A. The Protective Mechanism of SIRT1 in the Regulation of Mitochondrial Biogenesis and Mitochondrial Autophagy in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 149–157. [Google Scholar] [CrossRef]
- Rana, S.V. Metals and apoptosis: Recent developments. J. Trace Elem. Med. Biol. 2008, 22, 262–284. [Google Scholar] [CrossRef]
- Zhang, F.; Xing, S.; Li, Z. Antagonistic effects of lycopene on cadmium-induced hippocampal dysfunctions in autophagy, calcium homeostatis and redox. Oncotarget 2017, 8, 44720–44731. [Google Scholar] [CrossRef]
- Pi, H.; Li, M.; Tian, L.; Yang, Z.; Yu, Z.; Zhou, Z. Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci. Rep. 2017, 7, 43466. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Q.; Song, R.; Zhang, Y.; Yang, J.; Wang, Y.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; et al. Cadmium induced inhibition of autophagy is associated with microtubule disruption and mitochondrial dysfunction in primary rat cerebral cortical neurons. Neurotoxicol. Teratol. 2016, 53, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, S.; Xu, M.; Chen, X.; Wang, X.; Zhang, H.; Dong, X.; Zhang, R.; Chen, X.; Gao, W.; et al. Cadmium Impairs Autophagy Leading to Apoptosis by Ca2+-Dependent Activation of JNK Signaling Pathway in Neuronal Cells. Neurochem. Res. 2021, 46, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, S.; Goldstein, B. Sodium glucose co-transporter 2 inhibitors: Blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int. J. Clin. Pract. 2008, 62, 1279–1284. [Google Scholar] [CrossRef]
- Lee, T.-M.; Chang, N.-C.; Lin, S.-Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Wicinski, M.; Wodkiewicz, E.; Gorski, K.; Walczak, M.; Malinowski, B. Perspective of SGLT2 Inhibition in Treatment of Conditions Connected to Neuronal Loss: Focus on Alzheimer’s Disease and Ischemia-Related Brain Injury. Pharmaceuticals 2020, 13, 379. [Google Scholar] [CrossRef]
- Arab, H.H.; Safar, M.M.; Shahin, N.N. Targeting ROS-Dependent AKT/GSK-3beta/NF-kappaB and DJ-1/Nrf2 Pathways by Dapagliflozin Attenuates Neuronal Injury and Motor Dysfunction in Rotenone-Induced Parkinson’s Disease Rat Model. ACS Chem. Neurosci. 2021, 12, 689–703. [Google Scholar] [CrossRef] [PubMed]
- El-Sahar, A.E.; Rastanawi, A.A.; El-Yamany, M.F.; Saad, M.A. Dapagliflozin improves behavioral dysfunction of Huntington’s disease in rats via inhibiting apoptosis-related glycolysis. Life Sci. 2020, 257, 118076. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, R.N.; Ahmed, L.A.; Abdul Salam, R.M.; Ahmed, K.A.; Attia, A.S. Crosstalk Among NLRP3 Inflammasome, ETBR Signaling, and miRNAs in Stress-Induced Depression-Like Behavior: A Modulatory Role for SGLT2 Inhibitors. Neurotherapeutics 2021, 18, 2664–2681. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef]
- Anandhan, A.; Lei, S.; Levytskyy, R.; Pappa, A.; Panayiotidis, M.I.; Cerny, R.L.; Khalimonchuk, O.; Powers, R.; Franco, R. Glucose metabolism and AMPK signaling regulate dopaminergic cell death induced by gene (α-synuclein)-environment (paraquat) interactions. Mol. Neurobiol. 2017, 54, 3825–3842. [Google Scholar] [CrossRef] [PubMed]
- El-Kott, A.F.; Bin-Meferij, M.M.; Eleawa, S.M.; Alshehri, M.M. Kaempferol Protects Against Cadmium Chloride-Induced Memory Loss and Hippocampal Apoptosis by Increased Intracellular Glutathione Stores and Activation of PTEN/AMPK Induced Inhibition of Akt/mTOR Signaling. Neurochem. Res. 2020, 45, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Hussien, H.M.; Abd-Elmegied, A.; Ghareeb, D.A.; Hafez, H.S.; Ahmed, H.E.A.; El-Moneam, N.A. Neuroprotective effect of berberine against environmental heavy metals-induced neurotoxicity and Alzheimer’s-like disease in rats. Food Chem. Toxicol. 2018, 111, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Arab, H.H.; Gad, A.M.; Reda, E.; Yahia, R.; Eid, A.H. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: Targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021, 269, 119031. [Google Scholar] [CrossRef] [PubMed]
- Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Georgy, G.S. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals 2023, 16, 1065. [Google Scholar] [CrossRef]
- Arab, H.H.; Eid, A.H.; Yahia, R.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Saad, M.A.; Al-Shorbagy, M.Y.; Masoud, M.A. Targeting Autophagy, Apoptosis, and SIRT1/Nrf2 Axis with Topiramate Underlies Its Neuroprotective Effect against Cadmium-Evoked Cognitive Deficits in Rats. Pharmaceuticals 2023, 16, 1214. [Google Scholar] [CrossRef]
- Erdogan, M.A.; Yusuf, D.; Christy, J.; Solmaz, V.; Erdogan, A.; Taskiran, E.; Erbas, O. Highly selective SGLT2 inhibitor dapagliflozin reduces seizure activity in pentylenetetrazol-induced murine model of epilepsy. BMC Neurol. 2018, 18, 81. [Google Scholar] [CrossRef]
- Freireich, E.J.; Gehan, E.; Rall, D.; Schmidt, L.; Skipper, H. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother. Rep. 1966, 50, 219–244. [Google Scholar]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef]
- Wall, P.M.; Messier, C. Infralimbic kappa opioid and muscarinic M1 receptor interactions in the concurrent modulation of anxiety and memory. Psychopharmacology 2002, 160, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Arab, H.H.; Fikry, E.M.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Al-Hossaini, A.M.; Saad, M.A.; Al-Shorbagy, M.Y.; Eid, A.H. Stimulation of Autophagy by Dapagliflozin Mitigates Cadmium-Induced Testicular Dysfunction in Rats: The Role of AMPK/mTOR and SIRT1/Nrf2/HO-1 Pathways. Pharmaceuticals 2023, 16, 1006. [Google Scholar] [CrossRef] [PubMed]
- Arab, H.H.; Al-Shorbagy, M.Y.; Saad, M.A. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: Targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem. Biol. Interact. 2021, 335, 109368. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Q.; Huang, W.; Han, Y.; Tan, H.; An, M.; Xiang, Q.; Zhou, R.; Yang, L.; Cheng, Y. Dapagliflozin Alleviates Hepatic Steatosis by Restoring Autophagy via the AMPK-mTOR Pathway. Front. Pharmacol. 2021, 12, 589273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell. Signal. 2019, 55, 26–39. [Google Scholar] [CrossRef]
- Abd Elmaaboud, M.A.; Estfanous, R.S.; Atef, A.; Kabel, A.M.; Alnemari, K.A.; Naguib, T.M.; Alsufyani, S.E.; Darwish, H.W.; Arab, H.H. Dapagliflozin/Hesperidin Combination Mitigates Lipopolysaccharide-Induced Alzheimer’s Disease in Rats. Pharmaceuticals 2023, 16, 1370. [Google Scholar] [CrossRef]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef]
- Arab, H.H.; Elhemiely, A.A.; El-Sheikh, A.A.K.; Khabbaz, H.J.A.; Arafa, E.A.; Ashour, A.M.; Kabel, A.M.; Eid, A.H. Repositioning Linagliptin for the Mitigation of Cadmium-Induced Testicular Dysfunction in Rats: Targeting HMGB1/TLR4/NLRP3 Axis and Autophagy. Pharmaceuticals 2022, 15, 852. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, Y.; Zhang, M.; Deng, M.; Zhang, J.J. Intermittent Fasting Pretreatment Prevents Cognitive Impairment in a Rat Model of Chronic Cerebral Hypoperfusion. J. Nutr. 2017, 147, 1437–1445. [Google Scholar] [CrossRef]
- Babic, I.; Gorak, A.; Engel, M.; Sellers, D.; Else, P.; Osborne, A.L.; Pai, N.; Huang, X.F.; Nealon, J.; Weston-Green, K. Liraglutide prevents metabolic side-effects and improves recognition and working memory during antipsychotic treatment in rats. J. Psychopharmacol. 2018, 32, 578–590. [Google Scholar] [CrossRef]
- Iwamura, E.; Yamada, K.; Ichitani, Y. Involvement of hippocampal NMDA receptors in retrieval of spontaneous object recognition memory in rats. Behav. Brain Res. 2016, 307, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Waite, L.; Bonardi, C.; Stevenson, C.W.; Cassaday, H.J. Strain comparisons in inhibitory discrimination learning and novel object recognition procedures. Physiol. Behav. 2021, 240, 113557. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; He, X.; Wei, X.; Huang, J.; Zhang, J. After-effects of repetitive transcranial magnetic stimulation with parameter dependence on long-term potentiation-like plasticity and object recognition memory in rats. Front. Neurosci. 2023, 17, 1144480. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi-Tarie, R.; Kiasalari, Z.; Fakour, M.; Khorasani, M.; Keshtkar, S.; Baluchnejadmojarad, T.; Roghani, M. Nobiletin prevents amyloid beta(1-40)-induced cognitive impairment via inhibition of neuroinflammation and oxidative/nitrosative stress. Metab. Brain Dis. 2022, 37, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Millar, P.; Pathak, N.; Parthsarathy, V.; Bjourson, A.J.; O’Kane, M.; Pathak, V.; Moffett, R.C.; Flatt, P.R.; Gault, V.A. Metabolic and neuroprotective effects of dapagliflozin and liraglutide in diabetic mice. J. Endocrinol. 2017, 234, 255–267. [Google Scholar] [CrossRef]
- Zhang, Z.; Miah, M.; Culbreth, M.; Aschner, M. Autophagy in Neurodegenerative Diseases and Metal Neurotoxicity. Neurochem. Res. 2016, 41, 409–422. [Google Scholar] [CrossRef]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Silva, M.C.; Nandi, G.A.; Tentarelli, S.; Gurrell, I.K.; Jamier, T.; Lucente, D.; Dickerson, B.C.; Brown, D.G.; Brandon, N.J.; Haggarty, S.J. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 2020, 11, 3258. [Google Scholar] [CrossRef]
- Jaikumkao, K.; Promsan, S.; Thongnak, L.; Swe, M.T.; Tapanya, M.; Htun, K.T.; Kothan, S.; Intachai, N.; Lungkaphin, A. Dapagliflozin ameliorates pancreatic injury and activates kidney autophagy by modulating the AMPK/mTOR signaling pathway in obese rats. J. Cell Physiol. 2021, 236, 6424–6440. [Google Scholar] [CrossRef]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta 2006, 1762, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Weimann, A.; Simonsen, A.H.; Poulsen, H.E. Measurement of 8-oxo-7,8-dihydro-2’-deoxyguanosine and 8-oxo-7,8-dihydro-guanosine in cerebrospinal fluid by ultra performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2018, 1073, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dong, W.; Wang, R.; Li, Y.; Xu, B.; Zhang, J.; Zhao, Z.; Wang, Y. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res. Bull. 2015, 116, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant Activity of Fluoxetine and Vortioxetine in a Non-Transgenic Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Damri, O.; Shemesh, N.; Agam, G. Is There Justification to Treat Neurodegenerative Disorders by Repurposing Drugs? The Case of Alzheimer’s Disease, Lithium, and Autophagy. Int. J. Mol. Sci. 2021, 22, 189. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Back to Nucleus: Combating with Cadmium Toxicity Using Nrf2 Signaling Pathway as a Promising Therapeutic Target. Biol. Trace Elem. Res. 2020, 197, 52–62. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Received |
|---|---|---|
| Control | 10 | Group I received normal saline by gavage. Furthermore, animals received 0.5% carboxymethyl cellulose (CMC) by gavage. There was a two-hour interval between the two doses and the treatment protocol lasted for 2 months. |
| Control + DPG | 10 | Group II received normal saline by gavage. Furthermore, animals received dapagliflozin (1 mg/kg/day) by gavage. There was a two-hour interval between the two doses and the treatment protocol lasted for 2 months. |
| Cd | 10 | Group III received cadmium chloride (5 mg/kg/day) by gavage. Furthermore, animals received CMC by gavage. There was a two-hour interval between the two doses and the treatment protocol lasted for 2 months. The experimental regimen coincides with previously reported studies [22,23,24,25,26]. |
| Cd + DPG | 10 | Group IV received cadmium chloride (5 mg/kg/day) by gavage. Furthermore, animals received dapagliflozin (1 mg/kg/day) by gavage. There was a two-hour interval between the two doses and the treatment protocol lasted for 2 months. Prior studies were used to determine the dose of dapagliflozin as an effective dose for the attenuation of the behavioral deficits in Huntington-like manifestations [17], chronic stress-triggered depression-like behavior [19], rotenone-induced Parkinson’s disease [16], PTZ-induced epilepsy [27], and high-fat diet-fed rats [18]. Furthermore, dapagliflozin was administered in rats at a dose that is similar to the dosage regimen typically used in humans, according to the human equivalent dose (HED) calculation technique [28]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Sabry, F.M. Targeting Autophagy, Apoptosis, and Oxidative Perturbations with Dapagliflozin Mitigates Cadmium-Induced Cognitive Dysfunction in Rats. Biomedicines 2023, 11, 3000. https://doi.org/10.3390/biomedicines11113000
Arab HH, Eid AH, Alsufyani SE, Ashour AM, El-Sheikh AAK, Darwish HW, Sabry FM. Targeting Autophagy, Apoptosis, and Oxidative Perturbations with Dapagliflozin Mitigates Cadmium-Induced Cognitive Dysfunction in Rats. Biomedicines. 2023; 11(11):3000. https://doi.org/10.3390/biomedicines11113000
Chicago/Turabian StyleArab, Hany H., Ahmed H. Eid, Shuruq E. Alsufyani, Ahmed M. Ashour, Azza A. K. El-Sheikh, Hany W. Darwish, and Fatma M. Sabry. 2023. "Targeting Autophagy, Apoptosis, and Oxidative Perturbations with Dapagliflozin Mitigates Cadmium-Induced Cognitive Dysfunction in Rats" Biomedicines 11, no. 11: 3000. https://doi.org/10.3390/biomedicines11113000
APA StyleArab, H. H., Eid, A. H., Alsufyani, S. E., Ashour, A. M., El-Sheikh, A. A. K., Darwish, H. W., & Sabry, F. M. (2023). Targeting Autophagy, Apoptosis, and Oxidative Perturbations with Dapagliflozin Mitigates Cadmium-Induced Cognitive Dysfunction in Rats. Biomedicines, 11(11), 3000. https://doi.org/10.3390/biomedicines11113000