

Alphaviruses in Immunotherapy and Anticancer Therapy

Abstract
:1. Introduction
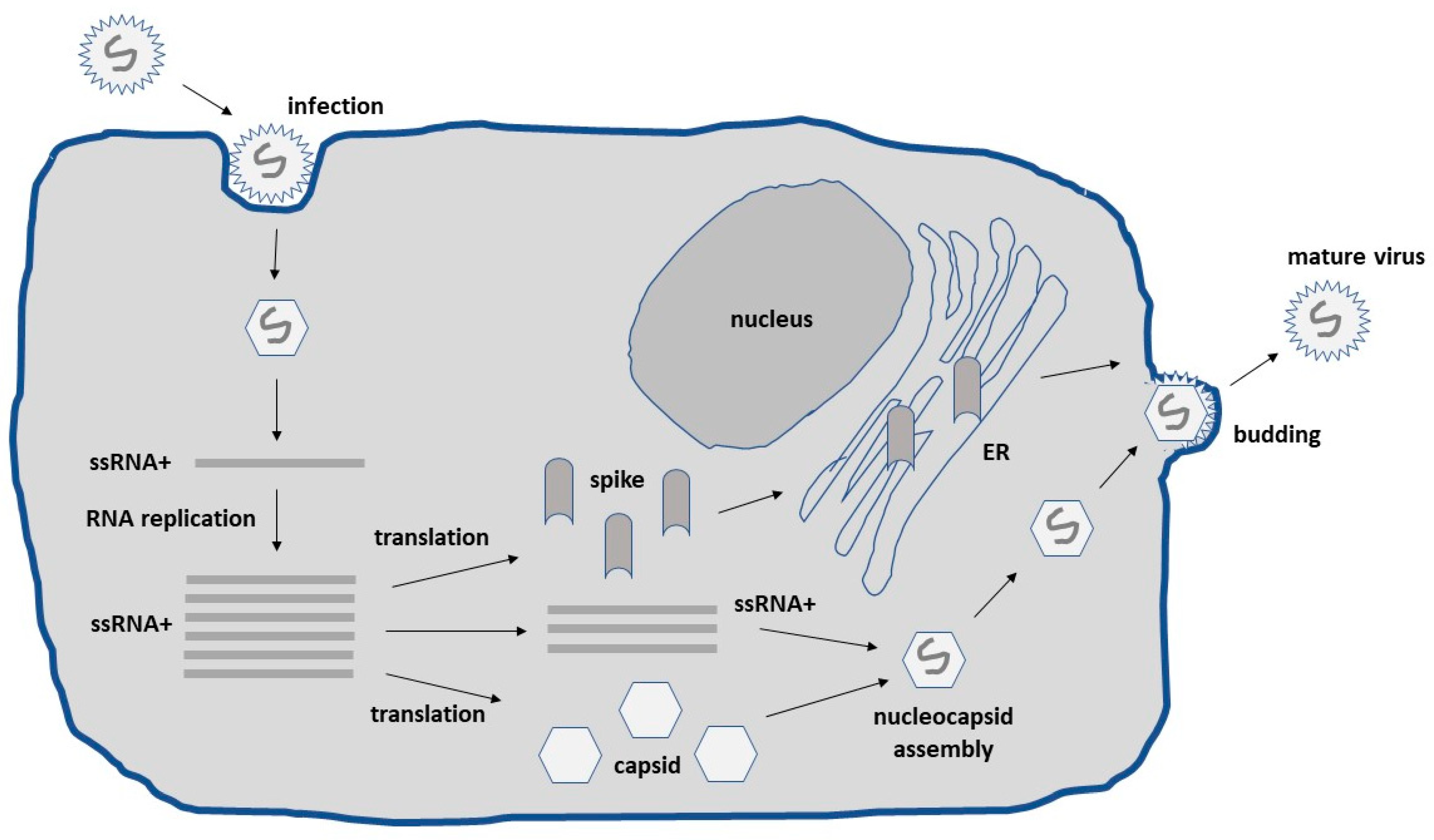
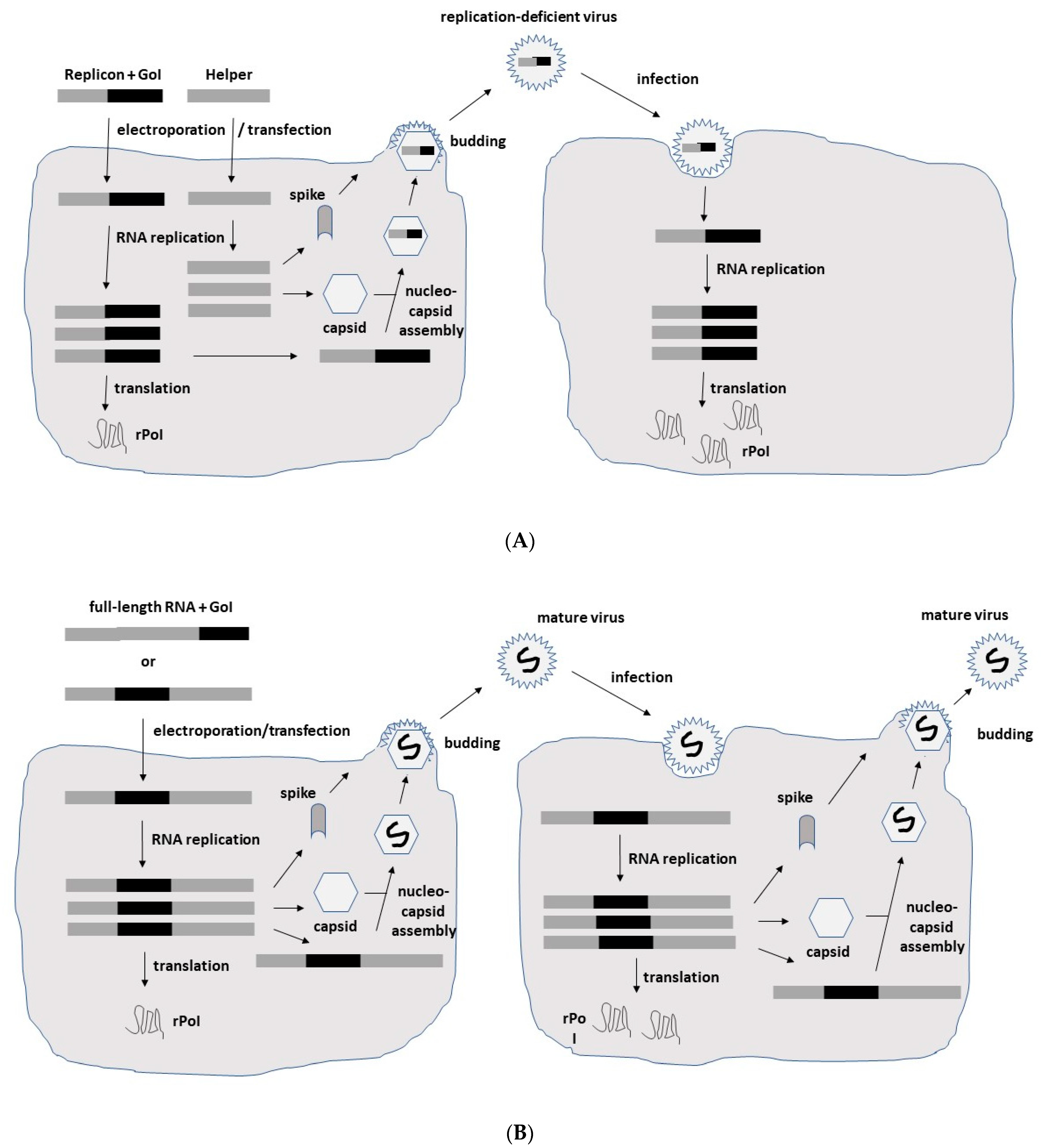
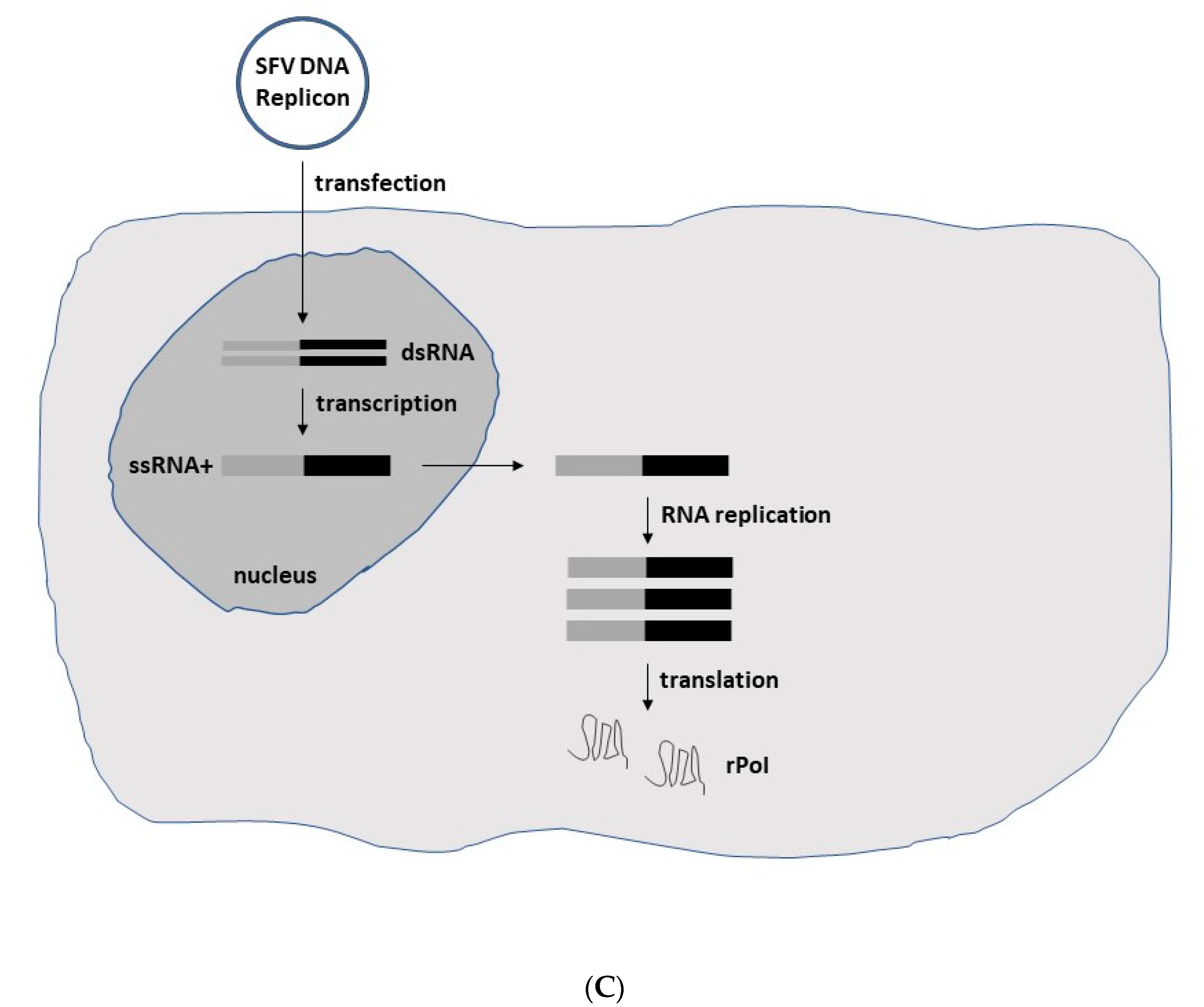
2. Alphavirus Lifecycle and Expression Vector Systems
3. Alphavirus-Based Immunotherapy for Cancer
3.1. Reporter Genes
3.2. Tumor-Associated Antigens
3.3. Cytotoxic and Antitumor Genes
3.4. Immunostimulatory Genes
3.5. Oncolytic Viruses
4. Conclusions
Funding
Conflicts of Interest
References
- Riley, R.S.; June, C.H.; Langer, R.; Mitchel, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Raja, J.; Ludwig, M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Alphaviruses in Cancer Therapy. Front. Mol. Biosci. 2022, 9, 864781. [Google Scholar] [CrossRef]
- Liljestrom, P.; Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Xiong, C.; Levis, R.; Shen, P.; Schlesinger, S.; Rice, C.M.; Huang, H.V. Sindbis virus: An efficient, broad host range vector for gene expression in animal cells. Science 1989, 243, 1188–1191. [Google Scholar] [CrossRef]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.F. In vitro synthesis of infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Tan, J.; Zhang, Y.; Wong, C.-W.; Lin, Z.; Liu, X.; Sander, M.; Yang, X.; Liang, L.; et al. Necroptotic Virotherapy of Oncolytic Alphavirus M1 Cooperated with Doxorubicin Displays Promising Therapeutic Efficacy in TNBC. Oncogene 2021, 40, 4783–4795. [Google Scholar] [CrossRef]
- Määttä, A.M.; Mäkinen, K.; Ketola, A.; Liimatainen, T.; Yongabi, F.N.; Vähä-Koskela, M. Replication Competent Semliki Forest Virus Prolongs Survival in Experimental Lung Cancer. Int. J. Cancer 2008, 123, 1704–1711. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Frolov, I.; Hoffman, T.A.; Pragal, B.M.; Dryga, S.A.; Huang, H.; Schlesinger, S.; Rice, C.M. Alphavirus-based expression vectors: Strategies and applications. Proc. Natl. Acad. Sci. USA 1996, 93, 11371–11377. [Google Scholar] [CrossRef] [Green Version]
- Frolova, E.; Frolov, I.; Schlesinger, S. Packaging Signals in Alphaviruses. J. Virol. 1997, 71, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. BNT162b2 mRNA COVID-19 vaccine: First approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.E.; Gargano, J.W.; Marin, M.; Wallace, M.; Curran, K.G.; Chamberland, M.; McClung, N.; Campos-Outcalt, D.; Morgan, R.L.; Mbaeyi, S.; et al. The advisory committee on immunization practices’ interim recommendation for use of Moderna COVID-19 vaccine—United States, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 69, 1653–1656. [Google Scholar] [CrossRef]
- DiCiommo, D.P.; Bremner, R. Rapid, high level protein production using DNA-based Semliki Forest virus vectors. J. Biol. Chem. 1998, 273, 18060–18066. [Google Scholar] [CrossRef]
- Olkkonen, V.M.; Liljestrom, P.; Garoff, H.; Simons, K.; Dotti, C.G. Expression of heterologous proteins in cultured rat hippocampal neurons using the Semliki Forest virus vector. J. Neurosc. Res. 1993, 35, 445–451. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Murphy, A.M.; Morris-Downes, M.M.; Sheahan, B.J.; Atkins, G.J. Inhibition of human lung carcinoma cell growth by apoptosis induction using Semliki Forest virus recombinant particles. Gene Ther. 2000, 7, 1477–1482. [Google Scholar] [CrossRef]
- Granot, T.; Yamanashi, Y.; Meruelo, D. Sindbis viral vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8+ T-cell immunity. Mol. Ther. 2014, 22, 112–122. [Google Scholar] [CrossRef]
- Ying, H.; Zaks, T.Z.; Wang, R.-F.; Irvine, K.R.; Kammula, U.S.; Marincola, F.M. Cancer therapy using a self-replicating RNA vaccine. Nat. Med. 1999, 5, 823–827. [Google Scholar] [CrossRef]
- Velders, M.P.; McElhiney, S.; Cassetti, M.C.; Eiben, G.L.; Higgins, T.; Kovacs, G.R. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001, 61, 7861–7867. [Google Scholar]
- Daemen, T.; Riezebos-Brilman, A.; Bungener, L.; Regts, J.; Dontje, B.; Wilschut, J. Eradication of established HPV16-transformed tumours after immunisation with recombinant Semliki Forest virus expressing a fusion protein of E6 and E7. Vaccine 2003, 21, 1082–1088. [Google Scholar] [CrossRef]
- Ip, P.P.; Boerma, A.; Walczak, M.; Oosterhuis, K.; Haanen, J.B.; Schumacher, T.N.; Nijman, H.W.; Daemen, T. Antigen design enhances the immunogenicity of Semliki Forest virus-based therapeutic human papillomavirus vaccines. Gene Ther. 2015, 22, 560–567. [Google Scholar] [CrossRef]
- Van de Wall, S.; Ljungberg, K.; Ip, P.P.; Boerma, A.; Knudsen, M.L.; Nijman, H.W.; Peter, L.; Daemen, T. Potent therapeutic efficacy of an alphavirus replicon DNA vaccine expressing human papilloma virus E6 and E7 antigens. Oncoimmunology 2018, 7, e1487913. [Google Scholar] [CrossRef]
- Komdeur, F.L.; Singh, A.; Van de Wall, S.; Meulenberg, J.J.M.; Boerma, A.; Hoogeboom, B.N.; Paijens, S.T.; Oyarce, C.; de Bruyn, M.; Schuuring, E.; et al. First-in-human phase I clinical trial of an SFV based RNA replicon cancer vaccine against HPV-induced cancers. Mol. Ther. 2021, 29, 611–625. [Google Scholar] [CrossRef]
- Lyons, J.A.; Sheahan, B.J.; Galbraith, S.E.; Mehra, R.; Atkins, G.J.; Fleeton, M.N. Inhibition of angiogenesis by a Semliki Forest virus vector expressing VEGFR-2 reduces tumour growth and metastasis in mice. Gene Ther. 2007, 14, 503–513. [Google Scholar] [CrossRef]
- Crosby, E.J.; Hobeika, A.C.; Niedzweicki, D.; Rushing, C.; Hsu, D.; Berglund, P.; Smith, J.; Osada, T.; Gwin, W.R., III; Hartman, Z.C.; et al. Long-term survival of patients with stage III colon cancer treated with VRP-CEA(6D), an alphavirus vector that increases the CD8+ effector memory T cell to Treg ration. J. Immunother. Cancer 2020, 8, e001662. [Google Scholar] [CrossRef]
- Morse, M.A.; Hobelka, A.C.; Osada, T.; Berglund, P.; Hubby, B.; Negri, S.; Niedzwiecki, D.; Devi, G.R.; Burnett, B.K.; Clay, T.M.; et al. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Investig. 2010, 120, 3234–3241. [Google Scholar] [CrossRef]
- Goldberg, S.M.; Bartido, S.M.; Gardner, J.P.; Guevara-Patino, J.A.; Montgomery, S.C.; Perales, M.-A.; Maughan, M.F.; Dempsey, J.; Donovan, G.P.; Olson, W.C.; et al. Comparison of two cancer vaccines targeting tyrosinase: Plamid DNA and recombinant alphavirus replicon particles. Clin. Cancer Res. 2005, 11, 8114–8121. [Google Scholar] [CrossRef]
- Avogadri, F.; Merghoub, T.; Maughan, M.F.; Hirschhorn-Cymerman, D.; Morris, J.; Ritter, E. Alphavirus replicon particles expressing TRP-2 provide potent therapeutic effect on melanoma through activation of humoral and cellular immunity. PLoS ONE 2010, 5, e12670. [Google Scholar] [CrossRef]
- Avogadri, F.; Zappasodi, R.; Yang, A.; Budhu, S.; Malandro, N.; Hisrchhorn-Cymerman, D. Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: Therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol. Res. 2014, 2, 448–458. [Google Scholar] [CrossRef]
- Yin, X.; Wang, W.; Zhu, X.; Wang, Y.; Wu, S.; Wang, Z. Synergistic antitumor efficacy of combined DNA vaccines targeting tumor cells and angiogenesis. Biochem. Biophys. Res. Comm. 2015, 465, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Tsai, Y.C.; Monie, A.; Wu, T.C.; Hung, C.F. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol. Ther. 2010, 18, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Durso, R.J.; Andjelic, S.; Gardner, J.P.; Margitich, D.J.; Donovan, G.P.; Arrigale, R.R.; Wang, X.; Maughan, M.F.; Talarico, T.L.; Olmsted, R.A.; et al. A novel alphavirus vaccine encoding prostate-specific membrane antigen elicits potent cellular and humoral immune responses. Clin. Cancer Res. 2007, 13, 3999–4008. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.F.; Kehoe, M.; Durso, R.; Fernandez, C.; Olson, W.; Gao, J.P.; Israel, R.; Scher, H.I.; Morris, S. A phase I dose escalation trial of vaccine replicon particles (VRP) expressing prostate-specific membrane antigen (PSMA) in subjects with prostate cancer. Vaccine 2013, 31, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Tretyakova, I.; Alexander, R.B.; Pushko, P.; Klyushnenkova, E.N. Anti-tumor effect of the alphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostate cancer. Vaccine 2015, 33, 5386–5395. [Google Scholar] [CrossRef]
- Garcia-Hernandez, M.L.; Gray, A.; Hubby, B.; Kast, W.M. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: A candidate antigen for treating prostate cancer. Cancer Res. 2007, 67, 1344–1351. [Google Scholar] [CrossRef]
- Garcia-Hernandez, M.L.; Gray, A.; Hubby, B.; Klinger, O.J.; Kast, W.M. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008, 68, 861–869. [Google Scholar] [CrossRef]
- Yamanaka, R.; Zullo, S.A.; Ramsey, J.; Onodera, M.; Tanaka, R.; Blaese, M. Induction of therapeutic antitumor anti-angiogenesis by intratumoral injection of genetically engineered endostatin-producing Semliki Forest virus. Cancer Gene Ther. 2001, 8, 796–802. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.P.; Rao, X.M.; Price, J.E.; Zhou, H.S.; Lachman, L.B. Prime-boost vaccination with plasmid and adenovirus gene vaccines control HER2/neu+ metastatic breast cancer in mice. Breast Cancer Res. 2005, 7, R580–R588. [Google Scholar] [CrossRef]
- Lachman, L.B.; Rao, X.M.; Kremer, R.H.; Ozpolat, B.; Kirjakova, G.; Price, J.E. DNA vaccination against neu reduces breast cancer incidence and metastasis in mice. Cancer Gene Ther. 2001, 8, 259–268. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.-P.; Maughan, M.F.; Lachman, L.B. Alphavirus replicon particles containing the gene for HER2/neu inhibit breast cancer growth and tumorigenesis. Breast Cancer Res. 2005, 7, R145–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosby, E.J.; Gwin, W.; Blackwell, K.; Marcom, P.K.; Chang, S.; Maecker, H.T.; Broadwater, G.; Hyslop, T.; Kim, S.; Rogatko, A.; et al. Vaccine-induced memory CD8(+) T cells provide clinical benefit in HER2 expressing breast cancer: A mouse to human translational study. Clin. Cancer Res. 2019, 25, 2725–2736. [Google Scholar] [CrossRef]
- Yamanaka, R.; Tsuchiya, N.; Yajima, N.; Honma, J.; Hasegawa, H.; Tanaka, R.; Ramsey, J.; Blaese, R.M.; Xanthopoulos, K.G. Induction of an antitumor immunological response by an intratumoral injection of dendritic cells pulsed with genetically engineered Semliki Forest virus to produce interleukin-18 combined with the systemic administration of interleukin-12. J. Neurosurg. 2003, 99, 746–753. [Google Scholar] [CrossRef]
- Roche, F.P.; Sheahan, B.J.; O’Mara, S.M.; Atkins, G.J. Semliki Forest virus-mediated gene therapy of the RG2 rat glioma. Neuropathol. Appl. Neurobiol. 2010, 36, 648–660. [Google Scholar] [CrossRef]
- Yamanaka, R.; Xanthopoulos, K.G. Induction of antigen-specific immune responses against malignant brain tumors by intramuscular injection of sindbis DNA encoding gp100 and IL-18. DNA Cell Biol. 2005, 24, 317–324. [Google Scholar] [CrossRef]
- Kramer, M.G.; Masner, M.; Casales, E.; Moreno, M.; Smerdou, C.; Chabalgoity, J.A. Neoadjuvant administration of Semliki Forest virus expressing interleukin-12 combined with attenuated Salmonella eradicates breast cancer metastasis and achieves long-term survival in immunocompetent mice. BMC Cancer 2015, 15, 620. [Google Scholar] [CrossRef]
- Rodriguez-Madoz, J.R.; Prieto, J.; Smerdou, C. Semliki Forest virus vectors engineered to express higher IL-12 levels induce efficient elimination of murine colon adenocarcinomas. Mol. Ther. 2005, 12, 153–163. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Labiano, S.; Aznar, M.A.; Bolanos, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sánchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef]
- Osada, T.; Berglund, P.; Morse, M.A.; Hubby, B.; Lewis, W.; Niedzwiecki, D.; Yang, X.Y.; Hobeika, A.; Burnett, B.; Devi, G.R.; et al. Co-delivery of antigen and IL-12 by Venezuelan equine encephalitis virus replicon particles enhances antigen-specific immune responses and anti-tumor effects. Cancer Immunol. Immunother. 2012, 61, 1941–1951. [Google Scholar] [CrossRef]
- Ren, H.; Boulikas, T.; Lundstrom, K.; Söling, P.C.; Warnke, P.C.; Rainov, N.G. Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki Forest virus vector carrying the human interleukin-12 gene—A phase I/II clinical protocol. J. Neurooncol. 2003, 64, 147–154. [Google Scholar] [CrossRef]
- Granot, T.; Meruelo, D. The role of natural killer cells in combinatorial anti-cancer therapy using Sindbis viral vector and irinotecan. Cancer Gene Ther. 2012, 19, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikkilä, J.E.; Vähä-Koskela, M.J.; Ruotsalainen, J.J.; Martikainen, M.W.; Stanford, M.M.; McCart, J.A.; Bell, J.C.; Hinkkanen, A.E. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS ONE 2010, 5, e8603. [Google Scholar] [CrossRef]
- Martikainen, M.; Ruotsalainen, J.; Tuomela, J.; Härkönen, P.; Essand, M.; Heikkilä, J.E.; Hinkkanen, A. Oncolytic alphavirus SFV-A7 efficiently eradicates subcutaneous and orthotopic prostate tumours in mice. Br. J. Cancer 2017, 117, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liang, J.; Tan, J.; Guo, L.; Cai, J.; Hu, J.; Yan, G.; Liu, Y.; Zhang, J.; Song, D.; et al. Real-Time Visualization and Quantification of Oncolytic M1 Virus In Vitro and In Vivo. Hum. Gene Ther. 2021, 32, 158–165. [Google Scholar] [CrossRef]
- Cai, J.; Zhu, W.; Lin, Y.; Zhang, S.; Chen, X.; Gong, S.; He, S.; Hu, J.; Yan, G.; Liang, J. Systematic Characterization of the Biodistribution of the Oncolytic Virus M1. Hum. Gene Ther. 2020, 31, 1203–1213. [Google Scholar] [CrossRef]
- Hu, C.; Liu, Y.; Lin, Y.; Liang, J.K.; Zhong, W.W.; Li, K.; Huang, W.; Wang, D.; Yan, G.; Zhu, W.; et al. Intravenous injections of the oncolytic virus M1 as a novel therapy for muscle-invasive bladder cancer. Cell Death Dis. 2018, 9, 274. [Google Scholar] [CrossRef]
- Sun, S.; Liu, Y.; He, C.; Hu, W.; Liu, W.; Huang, X.; Wu, J.; Xie, F.; Chen, C.; Wang, J.; et al. Combining Nanoknife with M1 oncolytic virus enhances anticancer activity in pancreatic cancer. Cancer Lett. 2021, 502, 9–24. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.; Zou, H.; Tian, X.; Hu, J.; Qiu, P.; Hu, H.; Yan, G. Liposome Encapsulation of Oncolytic Virus M1 To Reduce Immunogenicity and Immune Clearance in Vivo. Mol. Pharm. 2019, 16, 779–785. [Google Scholar] [CrossRef]
- Unno, Y.; Shino, Y.; Kondo, F.; Igarashi, N.; Wang, G.; Shimura, R.; Yamaguchi, T.; Asano, T.; Saisho, H.; Sekiya, S.; et al. Oncolytic virotherapy for cervical and ovarian cancer cells by Sindbis virus strain AR339. Clin. Cancer Res. 2005, 11, 4553–4560. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, H.; Liang, J.; Li, K.; Zhu, W.; Fu, L.; Wang, F.; Zheng, X.; Shi, H.; Wu, S.; et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc. Natl. Acad. Sci. USA 2014, 111, E4504–E4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, J.; Nie, L.; Jia, X.; Xu, C.; Cai, J.; Lin, Y.; Hu, J.; Zhu, W.; Li, Y.; Chen, D.; et al. Visualization of the Oncolytic Alphavirus M1 Life Cycle in Cancer Cells. Virol. Sin. 2021, 36, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, K.; Zhu, W.B.; Zhang, H.; Huang, W.T.; Liu, X.C.; Lin, Y.; Cai, J.; Yan, G.; Qiu, J.; et al. Suppression of CCDC6 sensitizes tumor to oncolytic virus M1. Neoplasia 2021, 23, 158–168. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Cancer | Vector | Finding | Ref |
|---|---|---|---|
| Reporter Genes | |||
| Lung | SFV-EGFP | Tumor regression in mice | [17] |
| Colon | SIN-LacZ | Complete tumor remission | [18] |
| SFV-LacZ RNA | Tumor regression, protection | [19] | |
| TAAs | |||
| Cervical | VEE-HPV-16 E7 | Protection against tumor challenges in mice | [20] |
| SFVenh-HPV E6-E7 | Tumor eradication, long-lasting CTL in mice | [21] | |
| SFV-sHELP-E7SH | Tumor regression, protection in mice | [22] | |
| SFV-HPV E6-E7 DNA | 85% of immunized mice tumor-free | [23] | |
| SFVenh-HPV E6-E7 | Phase I: Immunogenicity in all patients | [24] | |
| Colon | SFV-VEGFR-2 | Inhibition of tumor growth, metastatic spread | [25] |
| SFV-VEGR-2 + SFV-IL-4 | Prolonged survival after coadministration | [25] | |
| VEE-CEA | Phase I: Ag-specific response, long-term survival | [26] | |
| Pancreatic | VEE-CEA | Phase I: Prolonged survival | [27] |
| Melanoma | VEE-TRP-2 + DNA | Superior to plasmid DNA vaccine in mice | [28] |
| VEE-TRP-2 | Humoral immune responses, protection in mice | [29] | |
| VEE-TRP-2 + CTLA-4 mAbs | Tumor regression in 50% of mice | [30] | |
| VEE-TRP-2 + GITR mAbs | Tumor regression in 90% of mice | [30] | |
| SFV-VEGFR-2/IL-12 DNA | Synergistic antitumor activity from combination of | [31] | |
| + SFV-Survivin/β-hCG DNA | DNA replicons | ||
| Ovarian | SFV-OVA + VV-OVA | Immune responses, enhanced antitumor activity | [32] |
| Prostate | VEE-PSMA | Th1-biased immune responses | [33] |
| VEE-PSMA | Phase I: Good safety, weak immunogenicity | [34] | |
| VEE-PSA | PSA-specific Abs, delay in tumor growth | [35] | |
| VEE-mSTEAP + pcDNA | Prolonged survival, tumor challenge protection | [36] | |
| VEE-PSCA | Long-term survival of mice | [37] | |
| Cytotoxic and Antitumor Genes | |||
| Glioblastoma | SFV–Endostatin | Tumor growth inhibition, reduced vascularization | [38] |
| Breast | SIN-HER2/neu DNA | Significant tumor growth inhibition, protection | [39] |
| SIN-HER2/neu DNA | 80% less DNA needed compared to plasmid DNA | [40] | |
| VEE-HER2/neu ECD/TM | Complete prevention of tumors in mice | [41] | |
| VEE-HER2/neu ECD/TM | Safe, PR in 1 patient, SD in 2 patients | [42] | |
| Immunostimulation | |||
| Glioblastoma | SFV-IL-18 + rec IL-12 | Superior therapeutic effect of combination | [43] |
| Glioma | SFV-IL-12 | 70–97% tumor volume reduction in rats | [44] |
| Brain | SIN-gp100 + SIN-IL-12 DNA | Superior antitumor activity, prolonged survival | [45] |
| Breast | SFV-IL-12 + LV101 | Superior antitumor activity of combination | [46] |
| Colon | SFVenh-IL-12 | Complete tumor regression, long-term survival | [47] |
| SFV-IL-12 + anti-PD1 | Superior combination therapy in mice | [48] | |
| VEE-IL-12 + VEE-CEA | Superior combination therapy in mice | [49] | |
| Melanoma | SFV-IL-12 + anti-PD1 | Superior combination therapy in mice | [48] |
| LSFV-IL-12 | Phase I: Good safety and tolerability | [50] | |
| Ovarian | SIN-IL12 + Irinotecan | Long-term survival in 35% of mice | [51] |
| Oncolytic Viruses | |||
| Glioblastoma | SFV-VA-EGFP | Long-term survival in 16 out of 17 mice | [52] |
| Prostate | SFV-VA-EGFP | Complete tumor eradication in mice | [53] |
| Lung | SFV-VA-EGFP | Long-term survival in mice | [8] |
| Liver | M1 | Liver tumor targeting in mice | [54] |
| Glioma | M1 | Replication in tumors | [55] |
| Bladder | M1 | Tumor growth inhibition, prolonged survival | [56] |
| Breast | M1 + Doxorubicin | Reduced tumor growth in mice | [7] |
| Pancreatic | M1 + IRE | Tumor growth inhibition, prolonged survival | [57] |
| Cervical | SIN AR339 | Regression of established tumors in mice | [58] |
| Ovarian | SIN AR399 | Ascites formation in metastasis mouse model | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundstrom, K. Alphaviruses in Immunotherapy and Anticancer Therapy. Biomedicines 2022, 10, 2263. https://doi.org/10.3390/biomedicines10092263
Lundstrom K. Alphaviruses in Immunotherapy and Anticancer Therapy. Biomedicines. 2022; 10(9):2263. https://doi.org/10.3390/biomedicines10092263
Chicago/Turabian StyleLundstrom, Kenneth. 2022. "Alphaviruses in Immunotherapy and Anticancer Therapy" Biomedicines 10, no. 9: 2263. https://doi.org/10.3390/biomedicines10092263
APA StyleLundstrom, K. (2022). Alphaviruses in Immunotherapy and Anticancer Therapy. Biomedicines, 10(9), 2263. https://doi.org/10.3390/biomedicines10092263