
Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ Stimulus
 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Patients and Blood Collection
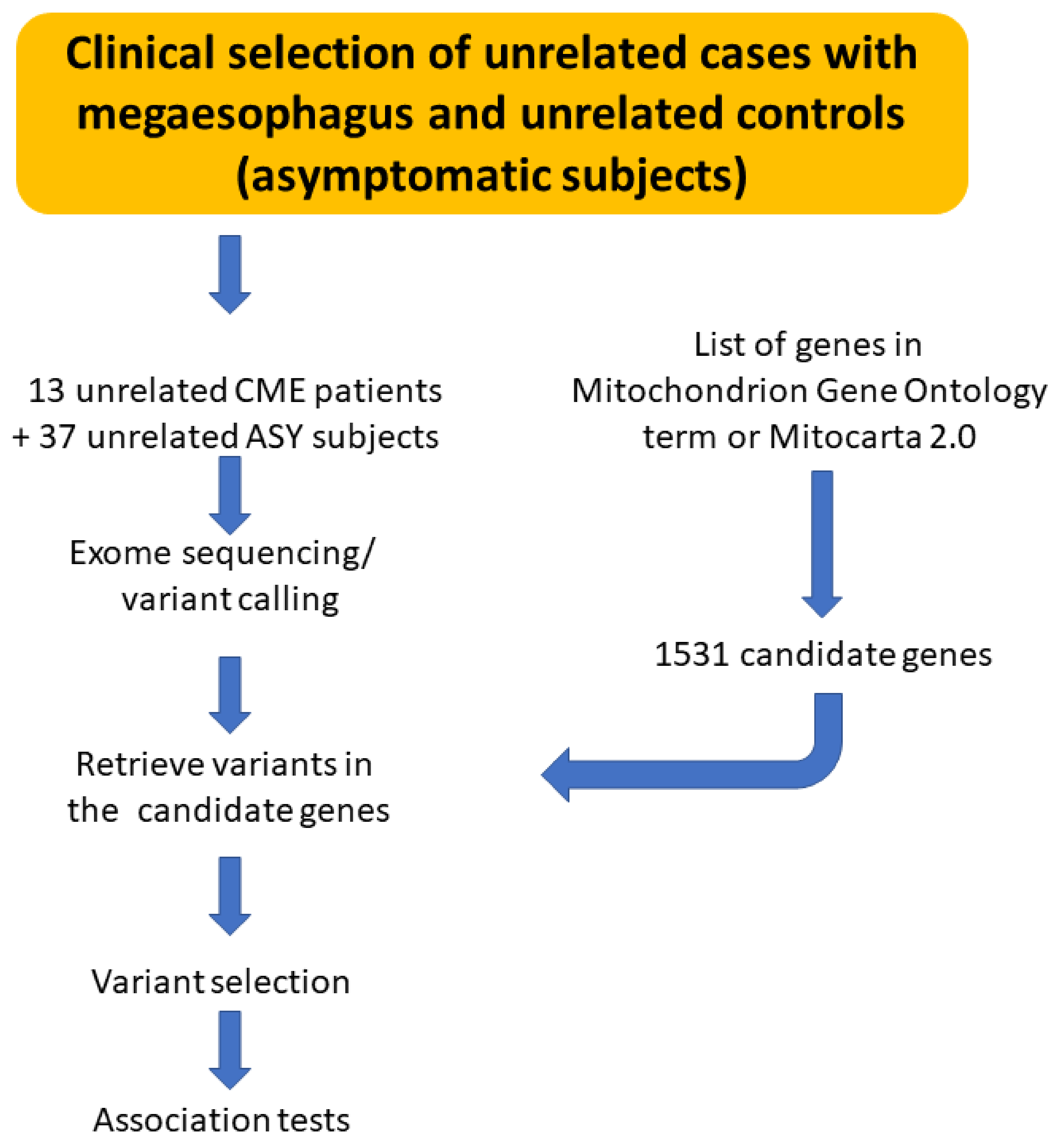
2.2. Whole Exome Sequencing (WES) of Patients with CME and ASY
2.3. Variants Selection
2.4. Isolation of Peripheral Blood Mononuclear Cells
2.5. Production of a Supernatant Rich in Epstein–Barr Virus Particles
2.6. Establishment of EBV-LCL from Patients
2.7. Evaluation of mtROS in LCLs
2.8. Sample Preparation of EBV-LCL Stimulated with IFN-γ
2.9. Quantification of Nitrite, ATP and Nitrated Protein
2.10. Statistical Analysis
3. Results
3.1. Exome Sequencing Reactions for CME Patients and ASY Subjects
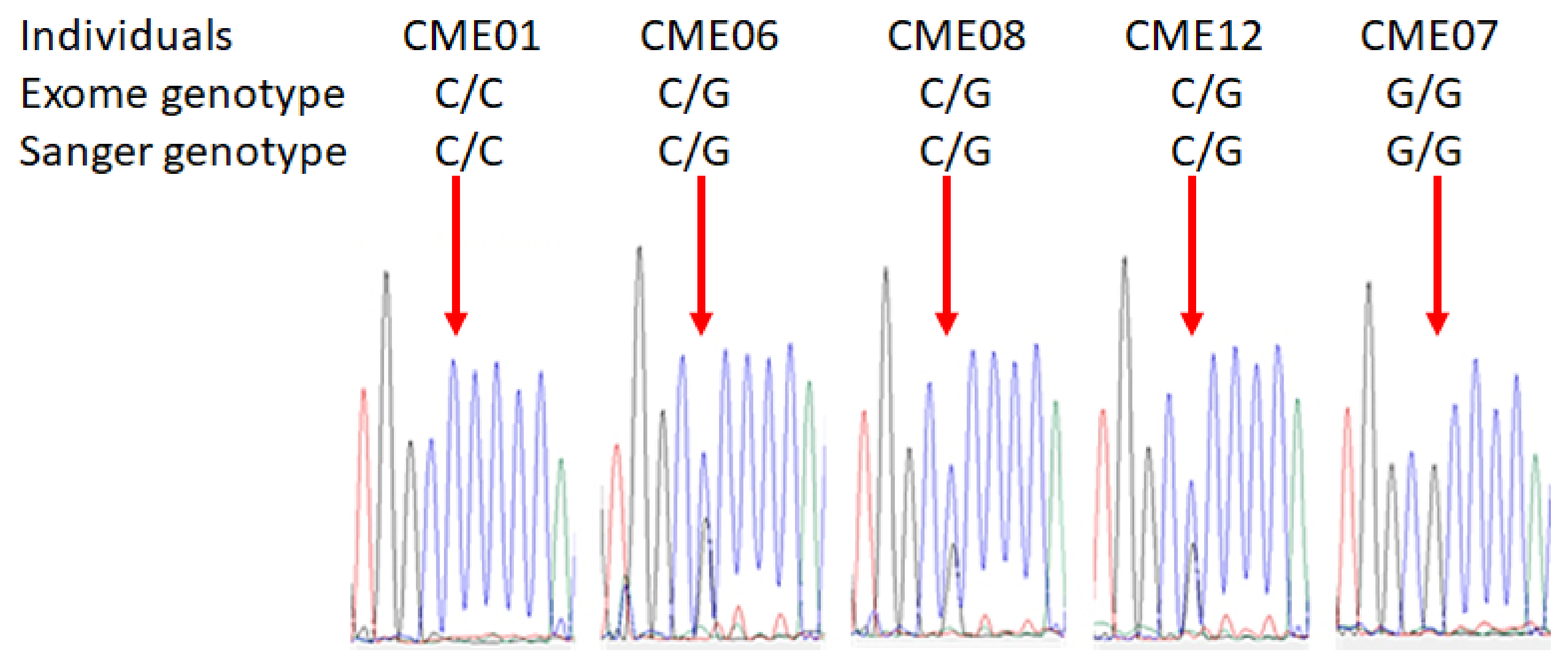
3.2. Pathogenic Variant of MRPS18B Is Accumulated in CME Patients
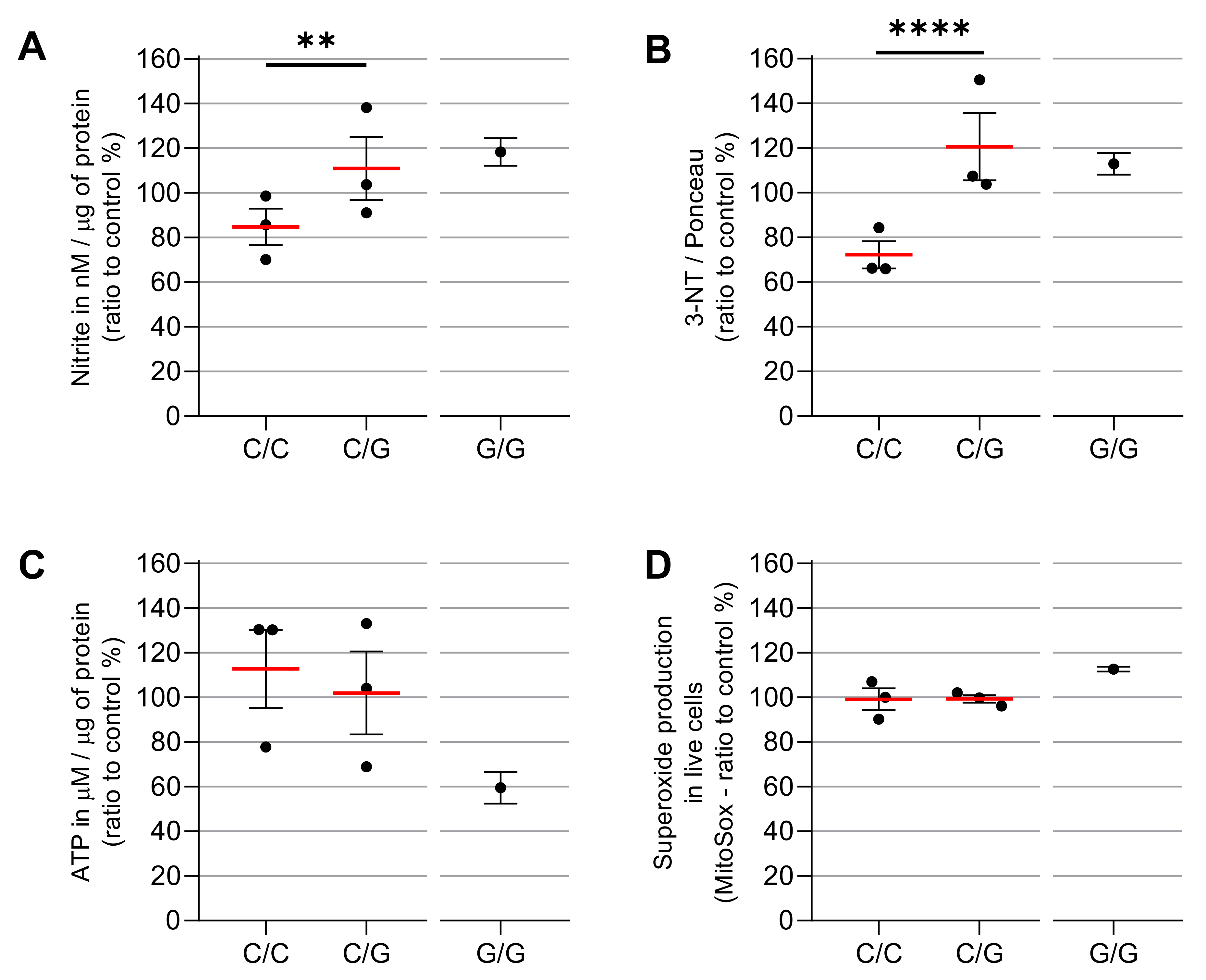
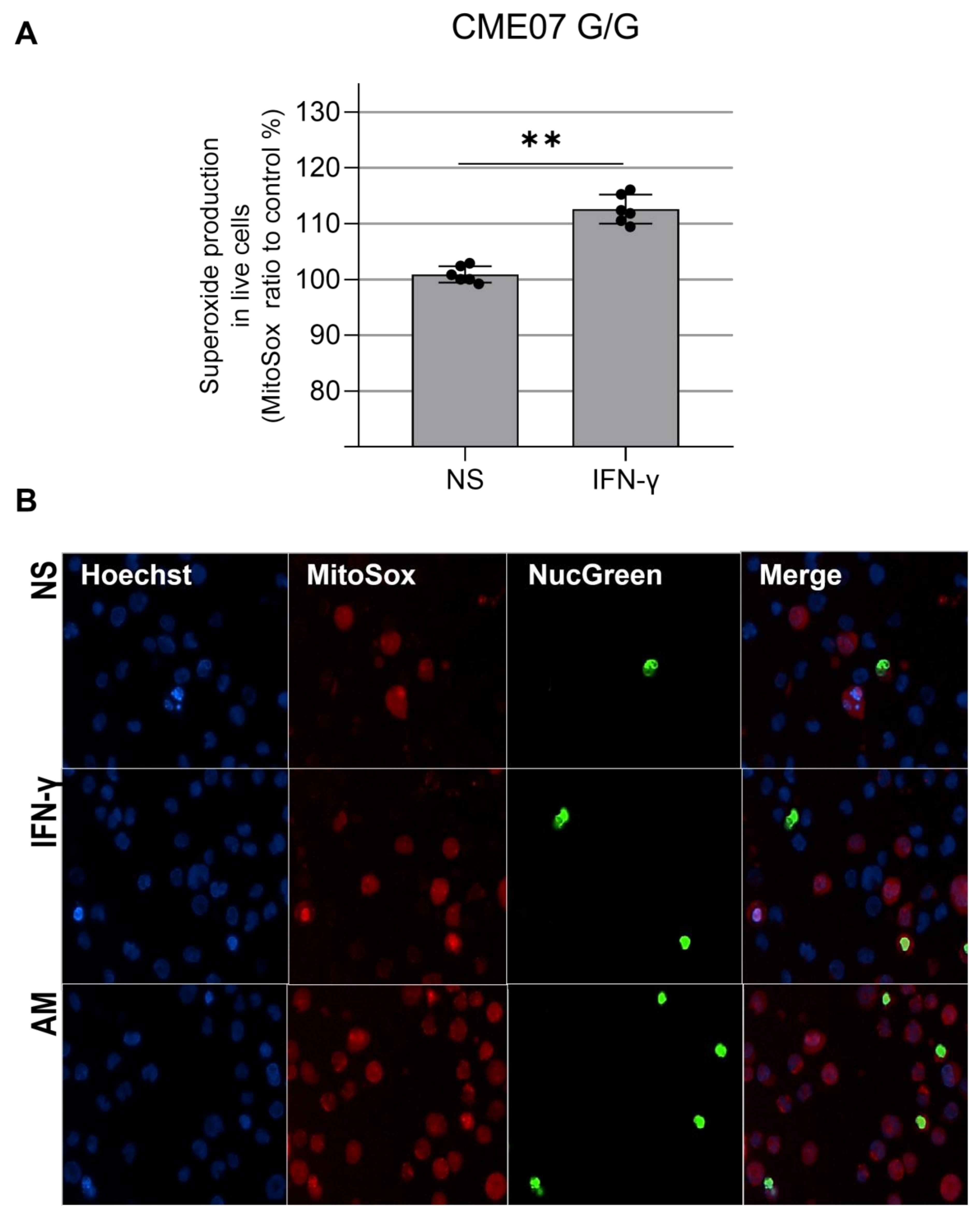
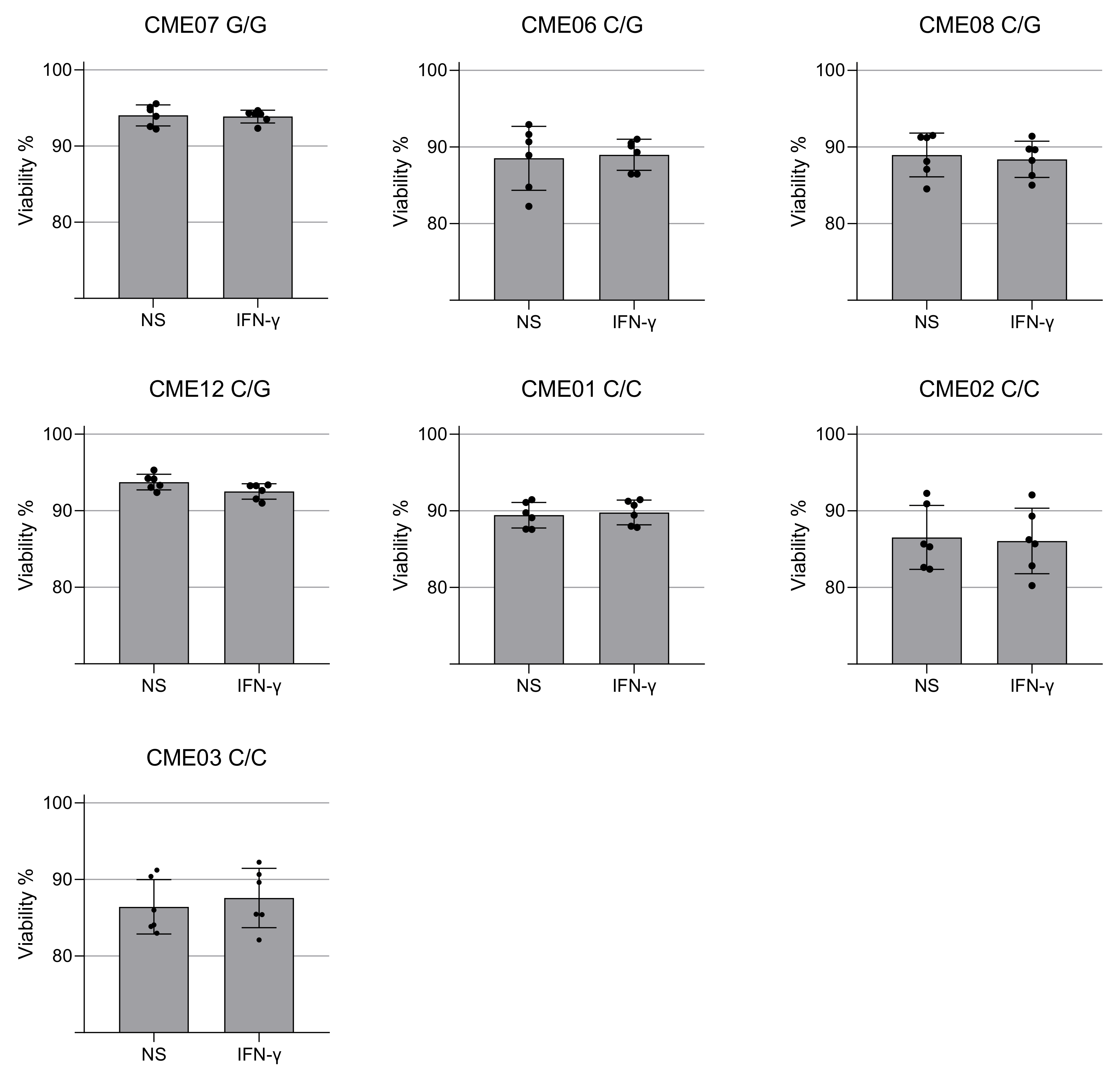
3.3. IFN-γ Causes Nitro-Oxidative Stress and Mitochondrial Damage in Patients Carrying MRPS18B P260A Variation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thakare, R.; Dasgupta, A.; Chopra, S. An update on benznidazole for the treatment of patients with Chagas disease. Drugs Today 2018, 54, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Neto, E.; Chevillard, C. Chagas disease cardiomyopathy: Immunopathology and genetics. Mediat. Inflamm. 2014, 2014, 683230. [Google Scholar] [CrossRef]
- Bocchi, E.A.; Bestetti, R.B.; Scanavacca, M.I.; Cunha Neto, E.; Issa, V.S. Chronic Chagas Heart Disease Management: From Etiology to Cardiomyopathy Treatment. J. Am. Coll. Cardiol. 2017, 70, 1510–1524. [Google Scholar] [CrossRef]
- Teixeira, A.R.; Nascimento, R.J.; Sturm, N.R. Evolution and pathology in chagas disease—a review. Mem. Inst. Oswaldo Cruz 2006, 101, 463–491. [Google Scholar] [CrossRef]
- Teixeira, A.R.; Nitz, N.; Guimaro, M.C.; Gomes, C.; Santos-Buch, C.A. Chagas disease. Postgrad. Med. J. 2006, 82, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.M.; Miller, S.M.; Evora, P.R. The chronic gastrointestinal manifestations of Chagas disease. Clinics 2009, 64, 1219–1224. [Google Scholar] [CrossRef]
- Zicker, F.; Smith, P.G.; Netto, J.C.; Oliveira, R.M.; Zicker, E.M. Physical activity, opportunity for reinfection, and sibling history of heart disease as risk factors for Chagas' cardiopathy. Am. J. Trop. Med. Hyg. 1990, 43, 498–505. [Google Scholar] [CrossRef]
- Chevillard, C.; Nunes, J.P.S.; Frade, A.F.; Almeida, R.R.; Pandey, R.P.; Nascimento, M.S.; Kalil, J.; Cunha-Neto, E. Disease Tolerance and Pathogen Resistance Genes May Underlie Trypanosoma cruzi Persistence and Differential Progression to Chagas Disease Cardiomyopathy. Front. Immunol. 2018, 9, 2791. [Google Scholar] [CrossRef] [PubMed]
- Luz, P.R.; Boldt, A.B.; Grisbach, C.; Kun, J.F.; Velavan, T.P.; Messias-Reason, I.J. Association of L-ficolin levels and FCN2 genotypes with chronic Chagas disease. PLoS ONE 2013, 8, e60237. [Google Scholar] [CrossRef]
- Dias, F.C.; Mendes-Junior, C.T.; Silva, M.C.; Tristao, F.S.; Dellalibera-Joviliano, R.; Moreau, P.; Soares, E.G.; Menezes, J.G.; Schmidt, A.; Dantas, R.O.; et al. Human leucocyte antigen-G (HLA-G) and its murine functional homolog Qa2 in the Trypanosoma cruzi Infection. Mediat. Inflamm. 2015, 2015, 595829. [Google Scholar] [CrossRef] [Green Version]
- Ouarhache, M.; Marquet, S.; Frade, A.F.; Ferreira, A.M.; Ianni, B.; Almeida, R.R.; Nunes, J.P.S.; Ferreira, L.R.P.; Rigaud, V.O.; Candido, D.; et al. Rare Pathogenic Variants in Mitochondrial and Inflammation-Associated Genes May Lead to Inflammatory Cardiomyopathy in Chagas Disease. J. Clin. Immunol. 2021, 41, 1048–1063. [Google Scholar] [CrossRef]
- Rossignol, R.; Malgat, M.; Mazat, J.P.; Letellier, T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 1999, 274, 33426–33432. [Google Scholar] [CrossRef]
- Finsterer, J. Mitochondriopathies. Eur. J. Neurol. 2004, 11, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Towbin, J.A.; Craigen, W.J.; Belmont, J.W.; Smith, E.O.; Neish, S.R.; Ware, S.M.; Hunter, J.V.; Fernbach, S.D.; Vladutiu, G.D.; et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef]
- Chelimsky, G.; Shanske, S.; Hirano, M.; Zinn, A.B.; Cohen, M.; McNeeley, K.; Chelimsky, T.C. Achalasia as the harbinger of a novel mitochondrial disorder in childhood. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 512–517. [Google Scholar] [CrossRef]
- Finsterer, J.; Frank, M. Gastrointestinal manifestations of mitochondrial disorders: A systematic review. Therap. Adv. Gastroenterol. 2017, 10, 142–154. [Google Scholar] [CrossRef]
- Chen, X.H.; Zhao, Y.P.; Xue, M.; Ji, C.B.; Gao, C.L.; Zhu, J.G.; Qin, D.N.; Kou, C.Z.; Qin, X.H.; Tong, M.L.; et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef]
- Lewis, J.A.; Huq, A.; Najarro, P. Inhibition of mitochondrial function by interferon. J. Biol. Chem. 1996, 271, 13184–13190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, B.M.; Crema, E.; Rodrigues, V., Jr. Analysis of the cellular immune response in patients with the digestive and indeterminate forms of Chagas' disease. Hum. Immunol. 2008, 69, 484–489. [Google Scholar] [CrossRef]
- Sousa, G.R.; Gomes, J.A.; Fares, R.C.; Damasio, M.P.; Chaves, A.T.; Ferreira, K.S.; Nunes, M.C.; Medeiros, N.I.; Valente, V.A.; Correa-Oliveira, R.; et al. Plasma cytokine expression is associated with cardiac morbidity in chagas disease. PLoS ONE 2014, 9, e87082. [Google Scholar] [CrossRef]
- Kalovidouris, A.E.; Plotkin, Z.; Graesser, D. Interferon-gamma inhibits proliferation, differentiation, and creatine kinase activity of cultured human muscle cells. II. A possible role in myositis. J. Rheumatol. 1993, 20, 1718–1723. [Google Scholar]
- Lee, H.J.; Oh, Y.K.; Rhee, M.; Lim, J.Y.; Hwang, J.Y.; Park, Y.S.; Kwon, Y.; Choi, K.H.; Jo, I.; Park, S.I.; et al. The role of STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial transmembrane potential during hepatic cell death induced by LPS/d-GalN. J. Mol. Biol. 2007, 369, 967–984. [Google Scholar] [CrossRef]
- Luss, H.; Watkins, S.C.; Freeswick, P.D.; Imro, A.K.; Nussler, A.K.; Billiar, T.R.; Simmons, R.L.; del Nido, P.J.; McGowan, F.X., Jr. Characterization of inducible nitric oxide synthase expression in endotoxemic rat cardiac myocytes in vivo and following cytokine exposure in vitro. J. Mol. Cell. Cardiol. 1995, 27, 2015–2029. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Laverny, G.; Allenbach, Y.; Grelet, E.; Ueberschlag, V.; Echaniz-Laguna, A.; Lannes, B.; Alsaleh, G.; Charles, A.L.; Singh, F.; et al. IFN-beta-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017, 134, 655–666. [Google Scholar] [CrossRef]
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNgamma-mediated metabolic reprogramming in cardiomyocytes. J. Pathol. 2019, 247, 320–332. [Google Scholar] [CrossRef]
- Wang, D.; McMillin, J.B.; Bick, R.; Buja, L.M. Response of the neonatal rat cardiomyocyte in culture to energy depletion: Effects of cytokines, nitric oxide, and heat shock proteins. Lab. Invest. 1996, 75, 809–818. [Google Scholar] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef]
- Geng, Y.; Hansson, G.K.; Holme, E. Interferon-gamma and tumor necrosis factor synergize to induce nitric oxide production and inhibit mitochondrial respiration in vascular smooth muscle cells. Circ. Res. 1992, 71, 1268–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bergen, N.J.; Crowston, J.G.; Kearns, L.S.; Staffieri, S.E.; Hewitt, A.W.; Cohn, A.C.; Mackey, D.A.; Trounce, I.A. Mitochondrial oxidative phosphorylation compensation may preserve vision in patients with OPA1-linked autosomal dominant optic atrophy. PLoS ONE 2011, 6, e21347. [Google Scholar] [CrossRef]
- Hussain, T.; Mulherkar, R. Lymphoblastoid Cell lines: A Continuous in Vitro Source of Cells to Study Carcinogen Sensitivity and DNA Repair. Int. J. Mol. Cell. Med. 2012, 1, 75–87. [Google Scholar]
- Castagna, A.E.; Addis, J.; McInnes, R.R.; Clarke, J.T.; Ashby, P.; Blaser, S.; Robinson, B.H. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. Am. J. Med. Genet. A 2007, 143A, 808–816. [Google Scholar] [CrossRef]
- Harvey, N.R.; Voisin, S.; Lea, R.A.; Yan, X.; Benton, M.C.; Papadimitriou, I.D.; Jacques, M.; Haupt, L.M.; Ashton, K.J.; Eynon, N.; et al. Investigating the influence of mtDNA and nuclear encoded mitochondrial variants on high intensity interval training outcomes. Sci. Rep. 2020, 10, 11089. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Cavdar Koc, E.; Burkhart, W.; Blackburn, K.; Moseley, A.; Spremulli, L.L. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J. Biol. Chem. 2001, 276, 19363–19374. [Google Scholar] [CrossRef] [PubMed]
- Gopisetty, G.; Thangarajan, R. Mammalian mitochondrial ribosomal small subunit (MRPS) genes: A putative role in human disease. Gene 2016, 589, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, M.; Ali, R.H.; Kashuba, V.; Klein, G.; Kashuba, E. S18 family of mitochondrial ribosomal proteins: Evolutionary history and Gly132 polymorphism in colon carcinoma. Oncotarget 2016, 7, 55649–55662. [Google Scholar] [CrossRef]
- Ferrari, A.; Del’Olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Santos, R.H.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Bocchi, E.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Myocardial gene expression of T-bet, GATA-3, Ror-gammat, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: An essentially unopposed TH1-type response. Mediators Inflamm. 2014, 2014, 914326. [Google Scholar] [CrossRef]
- Reis, D.D.; Jones, E.M.; Tostes, S., Jr.; Lopes, E.R.; Gazzinelli, G.; Colley, D.G.; McCurley, T.L. Characterization of inflammatory infiltrates in chronic chagasic myocardial lesions: Presence of tumor necrosis factor-alpha+ cells and dominance of granzyme A+, CD8+ lymphocytes. Am. J. Trop. Med. Hyg. 1993, 48, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Abel, L.C.; Rizzo, L.V.; Ianni, B.; Albuquerque, F.; Bacal, F.; Carrara, D.; Bocchi, E.A.; Teixeira, H.C.; Mady, C.; Kalil, J.; et al. Chronic Chagas’ disease cardiomyopathy patients display an increased IFN-gamma response to Trypanosoma cruzi infection. J. Autoimmun. 2001, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.P.S.; Andrieux, P.; Brochet, P.; Almeida, R.R.; Kitano, E.; Honda, A.K.; Iwai, L.K.; Andrade-Silva, D.; Goudenege, D.; Alcantara Silva, K.D.; et al. Co-Exposure of Cardiomyocytes to IFN-gamma and TNF-alpha Induces Mitochondrial Dysfunction and Nitro-Oxidative Stress: Implications for the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Front. Immunol. 2021, 12, 755862. [Google Scholar] [CrossRef]
- Gupta, S.; Bhatia, V.; Wen, J.J.; Wu, Y.; Huang, M.H.; Garg, N.J. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic. Biol. Med. 2009, 47, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Pissetti, C.W.; Correia, D.; de Oliveira, R.F.; Llaguno, M.M.; Balarin, M.A.; Silva-Grecco, R.L.; Rodrigues, V., Jr. Genetic and functional role of TNF-alpha in the development Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 2011, 5, e976. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Gupta, S.; Zago, M.P.; Davidson, M.M.; Dousset, P.; Amoroso, A.; Garg, N.J. Defects of mtDNA replication impaired mitochondrial biogenesis during Trypanosoma cruzi infection in human cardiomyocytes and chagasic patients: The role of Nrf1/2 and antioxidant response. J. Am. Heart Assoc. 2012, 1, e003855. [Google Scholar] [CrossRef]
- Arantes, R.M.; Marche, H.H.; Bahia, M.T.; Cunha, F.Q.; Rossi, M.A.; Silva, J.S. Interferon-gamma-induced nitric oxide causes intrinsic intestinal denervation in Trypanosoma cruzi-infected mice. Am. J. Pathol. 2004, 164, 1361–1368. [Google Scholar] [CrossRef]
- Rakshit, S.; Chandrasekar, B.S.; Saha, B.; Victor, E.S.; Majumdar, S.; Nandi, D. Interferon-gamma induced cell death: Regulation and contributions of nitric oxide, cJun N-terminal kinase, reactive oxygen species and peroxynitrite. Biochim. Biophys. Acta 2014, 1843, 2645–2661. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Santos, R.H.; Fiorelli, A.I.; Bilate, A.M.; Benvenuti, L.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Selective decrease of components of the creatine kinase system and ATP synthase complex in chronic Chagas disease cardiomyopathy. PLoS Negl. Trop. Dis. 2011, 5, e1205. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P. Does more MnSOD mean more hydrogen peroxide? Anti-Cancer Agents Med. Chem. 2011, 11, 178–180. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Dias Duarte Machado, L.G.; Thiebaut, D.; Ouari, O.; et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J. Biol. Chem. 2018, 293, 10363–10380. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Ensembl Reference | Chromosome | Position | Ref Allele | Mutated Allele | rs Number | Nucleotide Change (Transcript) | Amino Acid Change (Transcript) | CADD Score |
|---|---|---|---|---|---|---|---|---|---|
| MRPS18B | ENSG00000204568 | Chr6 | 30593485 | C | G | rs34315095 | ENST00000259873: exon7 688C > G | ENST00000259873: P230A | 27.9 |
| FAM185A | ENSG00000222011 | Chr7 | 102417753 | T | G | rs201667800 | ENST00000409231: exon5 538T > G | ENST00000409231: Y180D | 26.0 |
| Mutations | CME Patients | ASY Subjects | ||||
|---|---|---|---|---|---|---|
| Heterozygote Patients | Homozygote Patients | Non-Mutated Patients | Heterozygote Subjects | Homozygote Subjects | Non-Mutated Subjects | |
| MRPS18B rs34315095 C/G | 4 | 1 | 8 | 1 | 0 | 36 |
| FAM185A rs201667800 T/G | 6 | 0 | 7 | 2 | 0 | 35 |
| Mutations | CME Patients | ASY Subjects | Fisher Exact Test p-Value | |||
|---|---|---|---|---|---|---|
| Mutated Chromosomes | Non Mutated Chromosomes | Mutated Chromosomes | Non-Mutated Chromosomes | |||
| MRPS18B rs34315095 C/G | 6 | 20 | 1 | 73 | 0.001105404 | |
| FAM185A rs201667800 T/G | 6 | 20 | 2 | 72 | 0.003611684 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, K.D.A.; Nunes, J.P.S.; Andrieux, P.; Brochet, P.; Almeida, R.R.; Kuramoto Takara, A.C.K.; Pereira, N.B.; Abel, L.; Cobat, A.; Zaniratto, R.C.F.; et al. Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ Stimulus. Biomedicines 2022, 10, 2215. https://doi.org/10.3390/biomedicines10092215
Silva KDA, Nunes JPS, Andrieux P, Brochet P, Almeida RR, Kuramoto Takara ACK, Pereira NB, Abel L, Cobat A, Zaniratto RCF, et al. Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ Stimulus. Biomedicines. 2022; 10(9):2215. https://doi.org/10.3390/biomedicines10092215
Chicago/Turabian StyleSilva, Karla Deysiree Alcântara, João Paulo Silva Nunes, Pauline Andrieux, Pauline Brochet, Rafael Ribeiro Almeida, Andréia Cristina Kazue Kuramoto Takara, Natalia Bueno Pereira, Laurent Abel, Aurelie Cobat, Ricardo Costa Fernandes Zaniratto, and et al. 2022. "Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ Stimulus" Biomedicines 10, no. 9: 2215. https://doi.org/10.3390/biomedicines10092215
APA StyleSilva, K. D. A., Nunes, J. P. S., Andrieux, P., Brochet, P., Almeida, R. R., Kuramoto Takara, A. C. K., Pereira, N. B., Abel, L., Cobat, A., Zaniratto, R. C. F., Levy, D., Bydlowski, S. P., Cecconello, I., Seguro, F. C. B. d. C., Kalil, J., Chevillard, C., & Cunha-Neto, E. (2022). Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ Stimulus. Biomedicines, 10(9), 2215. https://doi.org/10.3390/biomedicines10092215