
NFkB Pathway and Hodgkin Lymphoma


{kind=link}
{kind=link}
Abstract
:1. Introduction
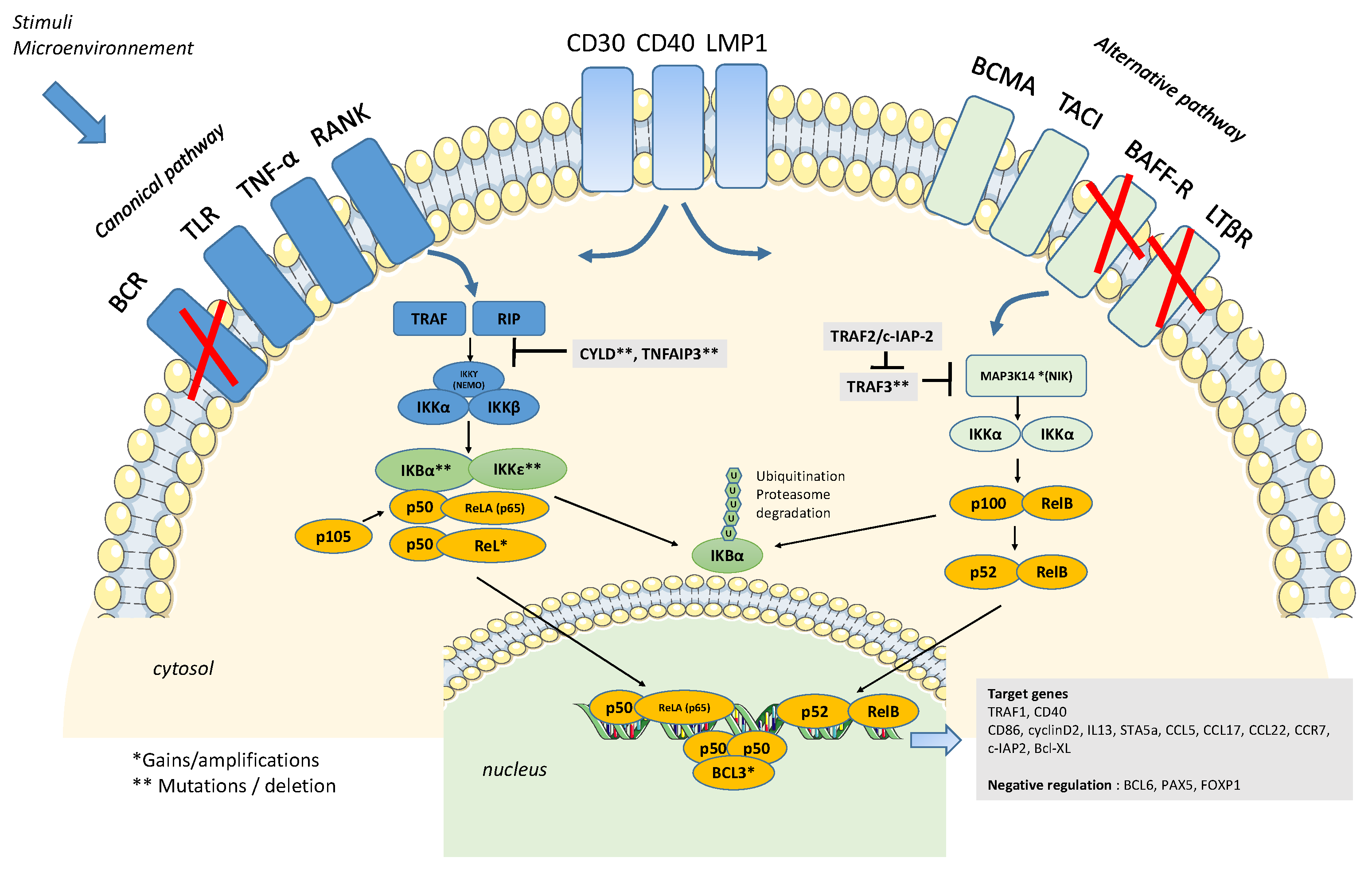
2. Constitutive Activation of the NFkB Pathway
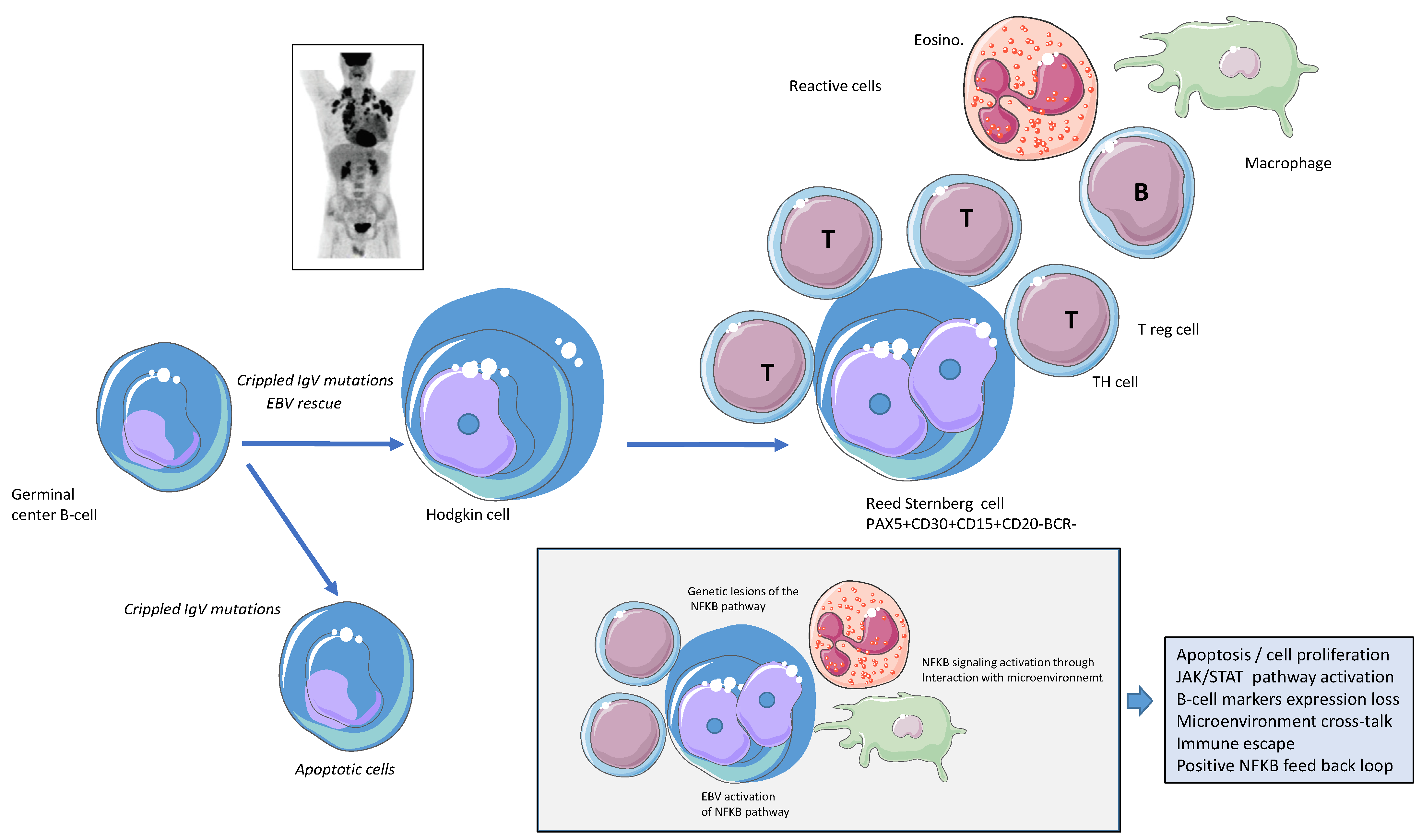
3. Activation of the NFK-B Pathway, the First Hit?
4. Somatic Mutations Related to NFkB Pathways/Genetic Lesions of Components of the NFkB Pathway
5. HL Microenvironment and NFkB Activation
6. Targeting the NFkB Pathway in cHL
7. Specificities of Nodular Lymphocyte Predominant HL
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Engert, A.; Younes, A. Hodgkin, Lymphoma: A Comprehensive Overview, 2nd ed.; Springer International Publising: Cham, Switzerland, 2015. [Google Scholar]
- Connors, J.M.; Cozen, W.; Steidl, C.; Carbone, A.; Hoppe, R.T.; Flechtner, H.H.; Bartlett, N.L. Hodgkin lymphoma. Nat. Rev. Dis. Primers 2020, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Kuppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Mottok, A.; Steidl, C. Biology of classical Hodgkin lymphoma: Implications for prognosis and novel therapies. Blood 2018, 131, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Kuppers, R. NF-kappaB deregulation in Hodgkin lymphoma. Semin. Cancer Biol. 2016, 39, 32–39. [Google Scholar] [CrossRef]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Mooller, P. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef]
- Rodig, S.J.; Savage, K.J.; Nguyen, V.; Pinkus, G.S.; Shipp, M.A.; Aster, J.C.; Kutok, J.L. TRAF1 expression and c-Rel activation are useful adjuncts in distinguishing classical Hodgkin lymphoma from a subset of morphologically or immunophenotypically similar lymphomas. Am. J. Surg. Pathol. 2005, 29, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Johrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Pittaluga, S.; Evbuomwan, M.O.; Jaffe, E.S.; Lewis, B.A. Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 2012, 120, 3756–3763. [Google Scholar] [CrossRef]
- de Oliveira, K.A.; Kaergel, E.; Heinig, M.; Fontaine, J.F.; Patone, G.; Muro, E.M.; Mathas, S.; Hummel, M.; Andrade-Navarro, M.A.; Hubner, N.; et al. A roadmap of constitutive NF-kappaB activity in Hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome Med. 2016, 8, 28. [Google Scholar] [CrossRef]
- Pittaluga, S.; Nicolae, A.; Wright, G.W.; Melani, C.; Roschewski, M.; Steinberg, S.; Huang, D.; Staudt, L.M.; Jaffe, E.S.; Wilson, W.H. Gene Expression Profiling of Mediastinal Gray Zone Lymphoma and Its Relationship to Primary Mediastinal B-cell Lymphoma and Classical Hodgkin Lymphoma. Blood Cancer Discov. 2020, 1, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Izban, K.F.; Ergin, M.; Huang, Q.; Qin, J.Z.; Martinez, R.L.; Schnitzer, B.; Ni, H.; Nickoloff, B.J.; Alkan, S. Characterization of NF-kappaB expression in Hodgkin’s disease: Inhibition of constitutively expressed NF-kappaB results in spontaneous caspase-independent apoptosis in Hodgkin and Reed-Sternberg cells. Mod. Pathol. 2001, 14, 297–310. [Google Scholar] [CrossRef]
- Hinz, M.; Loser, P.; Mathas, S.; Krappmann, D.; Dorken, B.; Scheidereit, C. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood 2001, 97, 2798–2807. [Google Scholar] [CrossRef]
- Gamboa-Cedeno, A.M.; Castillo, M.; Xiao, W.; Waldmann, T.A.; Ranuncolo, S.M. Alternative and canonical NF-kB pathways DNA-binding hierarchies networks define Hodgkin lymphoma and Non-Hodgkin diffuse large B Cell lymphoma respectively. J. Cancer Res. Clin. Oncol. 2019, 145, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R. B cells under influence: Transformation of B cells by Epstein-Barr virus. Nat. Rev. Immunol. 2003, 3, 801–812. [Google Scholar] [CrossRef]
- Kapatai, G.; Murray, P. Contribution of the Epstein Barr virus to the molecular pathogenesis of Hodgkin lymphoma. J. Clin. Pathol. 2007, 60, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Ferrari, A.; Ruffini, A.; Manzotti, G.; Fragliasso, V.; Merli, F.; Zanelli, M.; Valli, R.; Luminari, S.; Ciarrocchi, A. Gene expression profile unveils diverse biological effect of serum vitamin D in Hodgkin’s and diffuse large B-cell lymphoma. Hematol. Oncol. 2021, 39, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Sud, A.; Thomsen, H.; Orlando, G.; Forsti, A.; Law, P.J.; Broderick, P.; Cooke, R.; Hariri, F.; Pastinen, T.; Easton, D.F.; et al. Genome-wide association study implicates immune dysfunction in the development of Hodgkin lymphoma. Blood 2018, 132, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Sud, A.; Thomsen, H.; Law, P.J.; Forsti, A.; Filho, M.; Holroyd, A.; Broderick, P.; Orlando, G.; Lenive, O.; Wright, L.; et al. Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat. Commun. 2017, 8, 1892. [Google Scholar] [CrossRef] [Green Version]
- Gaiolla, R.D.; Moraes, M.P.T.; de Oliveira, D.E. SNPs in genes encoding for IL-10, TNF-alpha, and NFkappaB p105/p50 are associated with clinical prognostic factors for patients with Hodgkin lymphoma. PLoS ONE 2021, 16, e0248259. [Google Scholar] [CrossRef] [PubMed]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019, 3, 4065–4080. [Google Scholar] [CrossRef] [PubMed]
- Brune, M.M.; Juskevicius, D.; Haslbauer, J.; Dirnhofer, S.; Tzankov, A. Genomic Landscape of Hodgkin Lymphoma. Cancers 2021, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Szymanowska, N.; Klapper, W.; Gesk, S.; Kuppers, R.; Martin-Subero, J.I.; Siebert, R. BCL2 and BCL3 are recurrent translocation partners of the IGH locus. Cancer Genet. Cytogenet. 2008, 186, 110–114. [Google Scholar] [CrossRef]
- Martin-Subero, J.I.; Wlodarska, I.; Bastard, C.; Picquenot, J.M.; Hoppner, J.; Giefing, M.; Klapper, W.; Siebert, R. Chromosomal rearrangements involving the BCL3 locus are recurrent in classical Hodgkin and peripheral T-cell lymphoma. Blood 2006, 108, 401–402; author reply 402–403. [Google Scholar] [CrossRef]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef]
- Jungnickel, B.; Staratschek-Jox, A.; Brauninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.L.; Rajewsky, K.; Kuppers, R. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin’s lymphoma. J. Exp. Med. 2000, 191, 395–402. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Leveque, E.; Viailly, P.J.; Tonnelet, D.; Veresezan, E.L.; Drieux, F.; Etancelin, P.; Dubois, S.; Stamatoullas, A.; et al. Circulating tumor DNA in primary mediastinal large B-cell lymphoma versus classical Hodgkin lymphoma: A retrospective study. Leuk. Lymphoma 2022, 63, 834–844. [Google Scholar] [CrossRef]
- Schmidt, A.; Schmitz, R.; Giefing, M.; Martin-Subero, J.I.; Gesk, S.; Vater, I.; Massow, A.; Maggio, E.; Schneider, M.; Hansmann, M.L.; et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer 2010, 49, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Hung, S.S.; Chavez, E.A.; Duns, G.; Takata, K.; Chong, L.C.; Aoki, T.; Jiang, A.; Miyata-Takata, T.; Telenius, A.; et al. Mutational landscape of gray zone lymphoma. Blood 2021, 137, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Noerenberg, D.; Young, E.; Mylonas, E.; Abdulla, M.; Frick, M.; Asmar, F.; Ljungstrom, V.; Schneider, M.; Yoshida, K.; et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood 2016, 128, 2666–2670. [Google Scholar] [CrossRef]
- Mansouri, L.; Sutton, L.A.; Ljungstrom, V.; Bondza, S.; Arngarden, L.; Bhoi, S.; Larsson, J.; Cortese, D.; Kalushkova, A.; Plevova, K.; et al. Functional loss of IkappaBepsilon leads to NF-kappaB deregulation in aggressive chronic lymphocytic leukemia. J. Exp. Med. 2015, 212, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F.; Pujals, A.; Pelletier, L.; Bohers, E.; Camus, V.; Mareschal, S.; Dubois, S.; Sola, B.; Ochmann, M.; Lemonnier, F.; et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am. J. Hematol. 2016, 91, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Miloudi, H.; Taly, A.; Sola, B.; Jardin, F. XPO1 in B cell hematological malignancies: From recurrent somatic mutations to targeted therapy. J. Hematol. Oncol. 2017, 10, 47. [Google Scholar] [CrossRef]
- Taylor, J.; Sendino, M.; Gorelick, A.N.; Pastore, A.; Chang, M.T.; Penson, A.V.; Gavrila, E.I.; Stewart, C.; Melnik, E.M.; Herrejon Chavez, F.; et al. Altered Nuclear Export Signal Recognition as a Driver of Oncogenesis. Cancer Discov. 2019, 9, 1452–1467. [Google Scholar] [CrossRef]
- Nachmias, B.; Schimmer, A.D. Targeting nuclear import and export in hematological malignancies. Leukemia 2020, 34, 2875–2886. [Google Scholar] [CrossRef]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Desch, A.K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2021, 106, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Gruss, H.J.; Pinto, A. CD40 antigen expression on Reed-Sternberg cells. A reliable diagnostic tool for Hodgkin’s disease. Am. J. Pathol. 1995, 146, 780–781. [Google Scholar]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; De Paoli, P.; Zagonel, V.; Pinto, A. Expression of functional CD40 antigen on Reed-Sternberg cells and Hodgkin’s disease cell lines. Blood 1995, 85, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Safiran, Y.J.; Irving, S.G.; Kasid, U.N.; Cossman, J. Hodgkin disease: Pharmacologic intervention of the CD40-NF kappa B pathway by a protease inhibitor. Blood 2000, 96, 2841–2848. [Google Scholar] [CrossRef]
- Kilger, E.; Kieser, A.; Baumann, M.; Hammerschmidt, W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998, 17, 1700–1709. [Google Scholar] [CrossRef]
- Molin, D.; Fischer, M.; Xiang, Z.; Larsson, U.; Harvima, I.; Venge, P.; Nilsson, K.; Sundstrom, C.; Enblad, G.; Nilsson, G. Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin’s disease. Br. J. Haematol. 2001, 114, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.W.; et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007, 109, 729–739. [Google Scholar] [CrossRef]
- Szydłowski, M.; Prochorec-Sobieszek, M.; Szumera-Ciećkiewicz, A.; Derezińska, E.; Hoser, G.; Wasilewska, D.; Szymańska-Giemza, O.; Jabłońska, E.; Białopiotrowicz, E.; Sewastianik, T.; et al. Expression of PIM kinases in Reed-Sternberg cells fosters immune privilege and tumor cell survival in Hodgkin lymphoma. Blood 2017, 130, 1418–1429. [Google Scholar] [CrossRef]
- Toscano, M.A.; Campagna, L.; Molinero, L.L.; Cerliani, J.P.; Croci, D.O.; Ilarregui, J.M.; Fuertes, M.B.; Nojek, I.M.; Fededa, J.P.; Zwirner, N.W.; et al. Nuclear factor (NF)-kappaB controls expression of the immunoregulatory glycan-binding protein galectin-1. Mol. Immunol. 2011, 48, 1940–1949. [Google Scholar] [CrossRef]
- von Hoff, L.; Kargel, E.; Franke, V.; McShane, E.; Schulz-Beiss, K.W.; Patone, G.; Schleussner, N.; Kolesnichenko, M.; Hubner, N.; Daumke, O.; et al. Autocrine LTA signaling drives NF-kappaB and JAK-STAT activity and myeloid gene expression in Hodgkin lymphoma. Blood 2019, 133, 1489–1494. [Google Scholar] [CrossRef]
- Mohty, R.; Dulery, R.; Bazarbachi, A.H.; Savani, M.; Hamed, R.A.; Bazarbachi, A.; Mohty, M. Latest advances in the management of classical Hodgkin lymphoma: The era of novel therapies. Blood Cancer J. 2021, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Radford, J.; Connors, J.M.; Długosz-Danecka, M.; Kim, W.S.; Gallamini, A.; Ramchandren, R.; Friedberg, J.W.; Advani, R.; Hutchings, M.; et al. Overall Survival with Brentuximab Vedotin in Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the Involvement of the Master Transcription Factor NF-kappaB in Cancer Initiation and Progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liang, H.; Rao, E.; Zheng, W.; Huang, X.; Deng, L.; Zhang, Y.; Yu, X.; Xu, M.; Mauceri, H.; et al. Non-canonical NF-κB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e494. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; He, Y.; Tang, H.; Chen, X.; Liu, S.; Tao, Y. cGAS/STING: Novel perspectives of the classic pathway. Mol. Biomed. 2020, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]
- Blum, K.A.; Johnson, J.L.; Niedzwiecki, D.; Canellos, G.P.; Cheson, B.D.; Bartlett, N.L. Single agent bortezomib in the treatment of relapsed and refractory Hodgkin lymphoma: Cancer and leukemia Group B protocol 50206. Leuk. Lymphoma 2007, 48, 1313–1319. [Google Scholar] [CrossRef]
- Mendler, J.H.; Kelly, J.; Voci, S.; Marquis, D.; Rich, L.; Rossi, R.M.; Bernstein, S.H.; Jordan, C.T.; Liesveld, J.; Fisher, R.I.; et al. Bortezomib and gemcitabine in relapsed or refractory Hodgkin’s lymphoma. Ann. Oncol. 2008, 19, 1759–1764. [Google Scholar] [CrossRef]
- Zheng, B.; Georgakis, G.V.; Li, Y.; Bharti, A.; McConkey, D.; Aggarwal, B.B.; Younes, A. Induction of cell cycle arrest and apoptosis by the proteasome inhibitor PS-341 in Hodgkin disease cell lines is independent of inhibitor of nuclear factor-kappaB mutations or activation of the CD30, CD40, and RANK receptors. Clin. Cancer Res. 2004, 10, 3207–3215. [Google Scholar] [CrossRef]
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009, 114, 1046–1052. [Google Scholar] [CrossRef]
- Hamadani, M.; Balasubramanian, S.; Hari, P.N. Ibrutinib in Refractory Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 373, 1381–1382. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Astle, J.; Kakar, I.K.; Zellner, K.; Hari, P.N.; Hamadani, M. Clinical activity of ibrutinib in classical Hodgkin lymphoma relapsing after allogeneic stem cell transplantation is independent of tumor BTK expression. Br. J. Haematol. 2020, 190, e98–e101. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Chaker, M.; Aboukameel, A.; Mohammad, R.M.; Azmi, A.S.; Ramchandren, R. Pre-clinical anti-tumor activity of Bruton’s Tyrosine Kinase inhibitor in Hodgkin’s Lymphoma cellular and subcutaneous tumor model. Heliyon 2019, 5, e02290. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Dorken, B.; Jundt, F. Notch is an essential upstream regulator of NF-kappaB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia 2012, 26, 806–813. [Google Scholar] [CrossRef]
- Guorgui, J.; Wang, R.; Mattheolabakis, G.; Mackenzie, G.G. Curcumin formulated in solid lipid nanoparticles has enhanced efficacy in Hodgkin’s lymphoma in mice. Arch. Biochem. Biophys. 2018, 648, 12–19. [Google Scholar] [CrossRef]
- Xiao, Z.; Su, Z.; Han, S.; Huang, J.; Lin, L.; Shuai, X. Dual pH-sensitive nanodrug blocks PD-1 immune checkpoint and uses T cells to deliver NF-kappaB inhibitor for antitumor immunotherapy. Sci. Adv. 2020, 6, eaay7785. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Betzler, A.C.; Theodoraki, M.N.; Schuler, P.J.; Döscher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C. NF-κB and Its Role in Checkpoint Control. Int. J. Mol. Sci. 2020, 21, 3949. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.J. Progress in understanding the biology of nodular lymphocyte-predominant Hodgkin lymphoma. Haematologica 2021, 106, 2538. [Google Scholar] [CrossRef]
- Hartmann, S.; Eichenauer, D.A. Nodular lymphocyte predominant Hodgkin lymphoma: Pathology, clinical course and relation to T-cell/histiocyte rich large B-cell lymphoma. Pathology 2020, 52, 142–153. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Schmitz, R.; Brune, V.; Tiacci, E.; Doring, C.; Hansmann, M.L.; Siebert, R.; Kuppers, R. Mutations in the genes coding for the NF-kappaB regulating factors IkappaBalpha and A20 are uncommon in nodular lymphocyte-predominant Hodgkin’s lymphoma. Haematologica 2010, 95, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, S.; Doring, C.; Vucic, E.; Chan, F.C.; Ennishi, D.; Tousseyn, T.; de Wolf-Peeters, C.; Perner, S.; Wlodarska, I.; Steidl, C.; et al. Array comparative genomic hybridization reveals similarities between nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma. Br. J. Haematol. 2015, 169, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Schuhmacher, B.; Rausch, T.; Fuller, L.; Doring, C.; Weniger, M.; Lollies, A.; Weiser, C.; Thurner, L.; Rengstl, B.; et al. Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia 2016, 30, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Thurner, L.; Hartmann, S.; Fadle, N.; Regitz, E.; Kemele, M.; Kim, Y.J.; Bohle, R.M.; Nimmesgern, A.; von Muller, L.; Kempf, V.A.J.; et al. Lymphocyte predominant cells detect Moraxella catarrhalis-derived antigens in nodular lymphocyte-predominant Hodgkin lymphoma. Nat. Commun. 2020, 11, 2465. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardin, F. NFkB Pathway and Hodgkin Lymphoma. Biomedicines 2022, 10, 2153. https://doi.org/10.3390/biomedicines10092153
Jardin F. NFkB Pathway and Hodgkin Lymphoma. Biomedicines. 2022; 10(9):2153. https://doi.org/10.3390/biomedicines10092153
Chicago/Turabian StyleJardin, Fabrice. 2022. "NFkB Pathway and Hodgkin Lymphoma" Biomedicines 10, no. 9: 2153. https://doi.org/10.3390/biomedicines10092153
APA StyleJardin, F. (2022). NFkB Pathway and Hodgkin Lymphoma. Biomedicines, 10(9), 2153. https://doi.org/10.3390/biomedicines10092153