
Integrated Bioinformatics Analysis of the Hub Genes Involved in Irinotecan Resistance in Colorectal Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Processing and Analysis
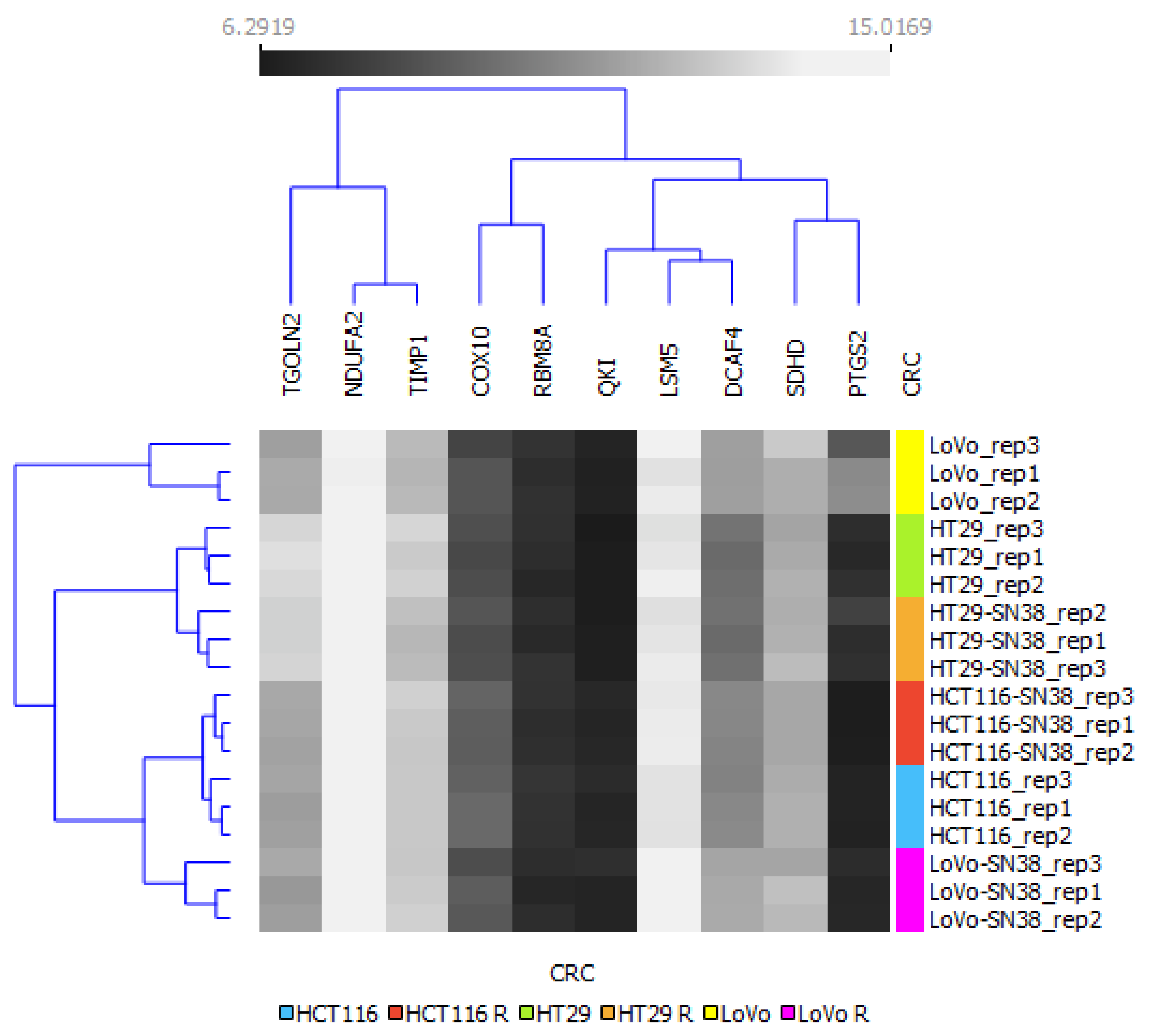
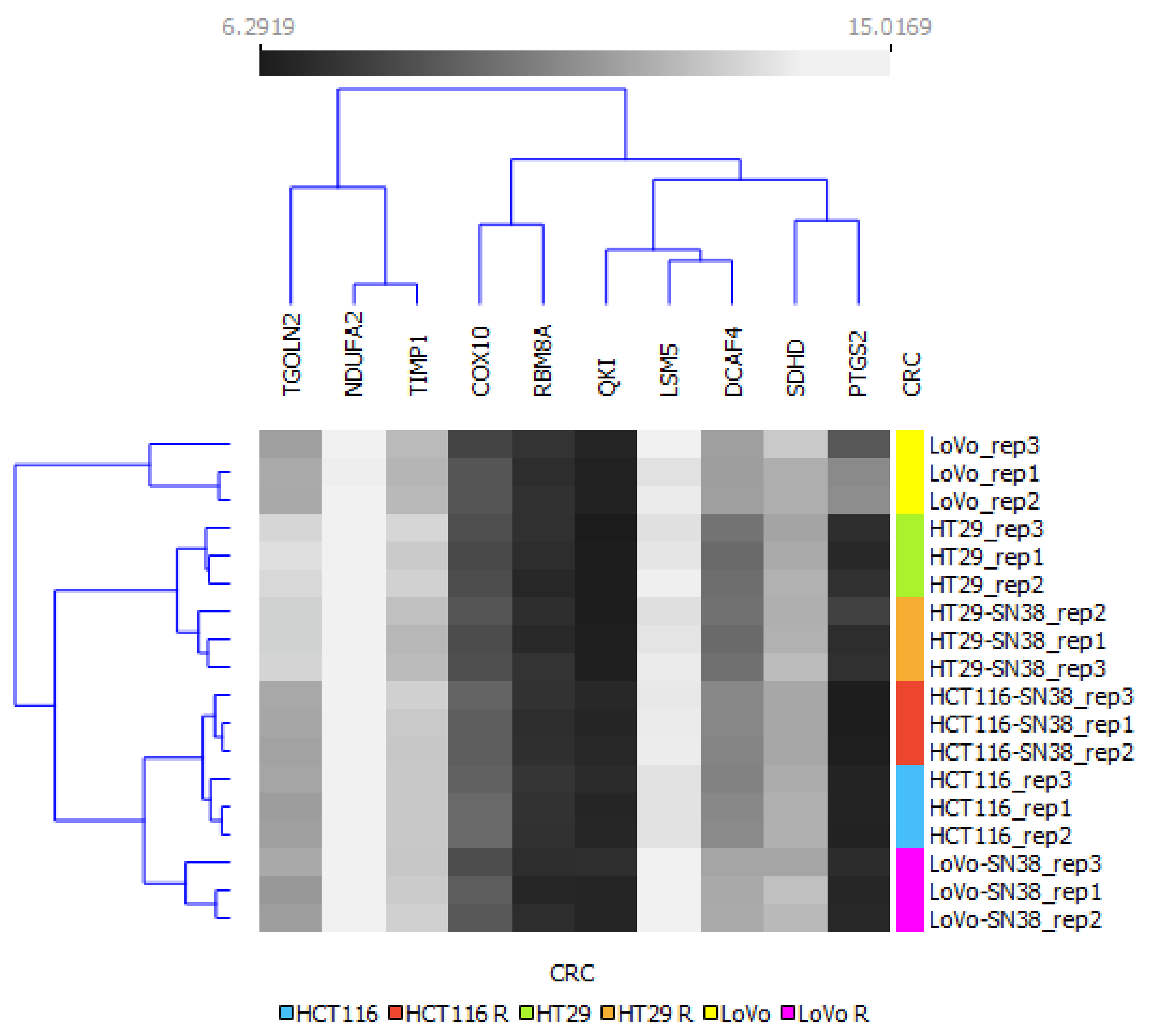
2.2. Hierarchical Clustering Analysis
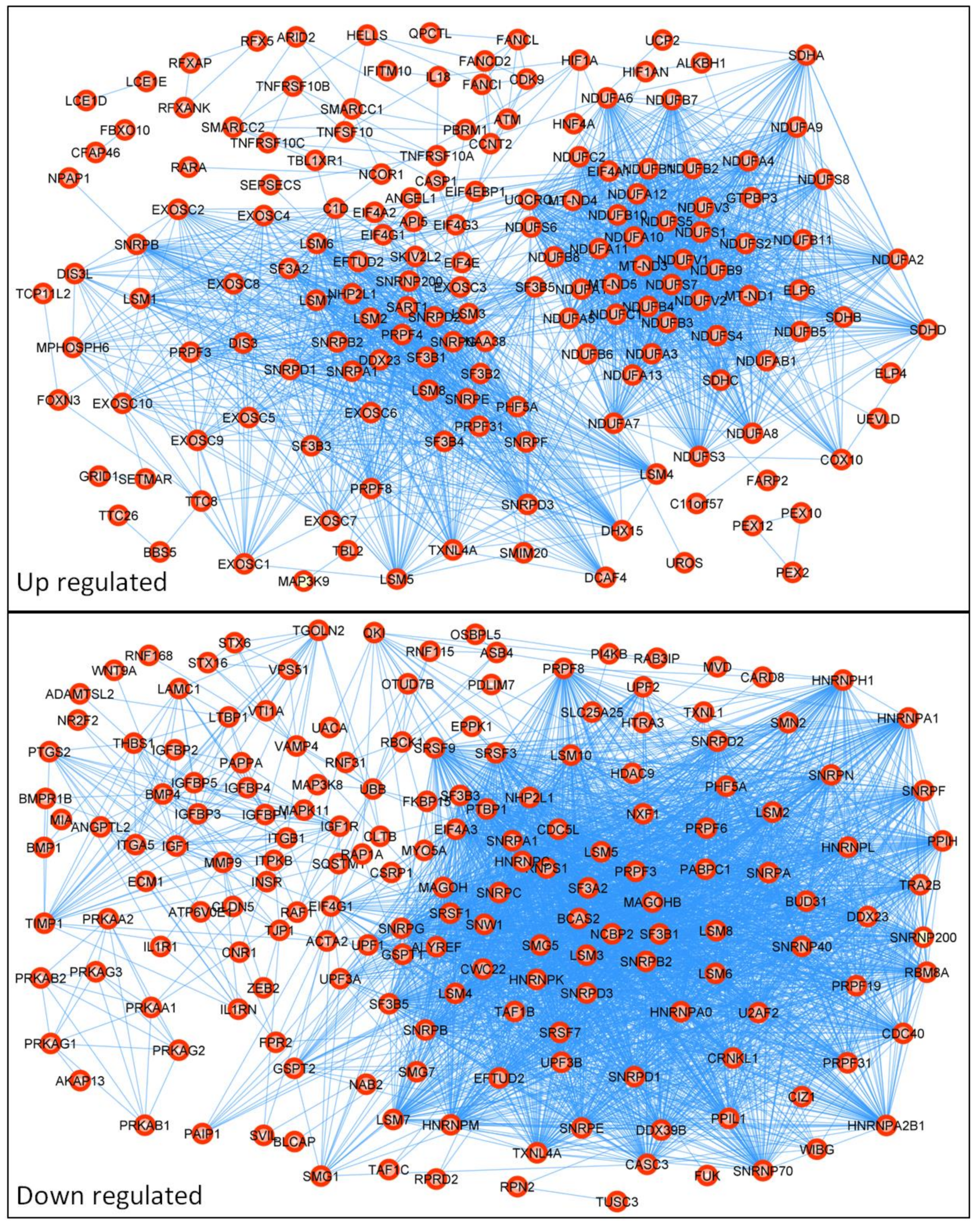
2.3. Construction of PPI Network
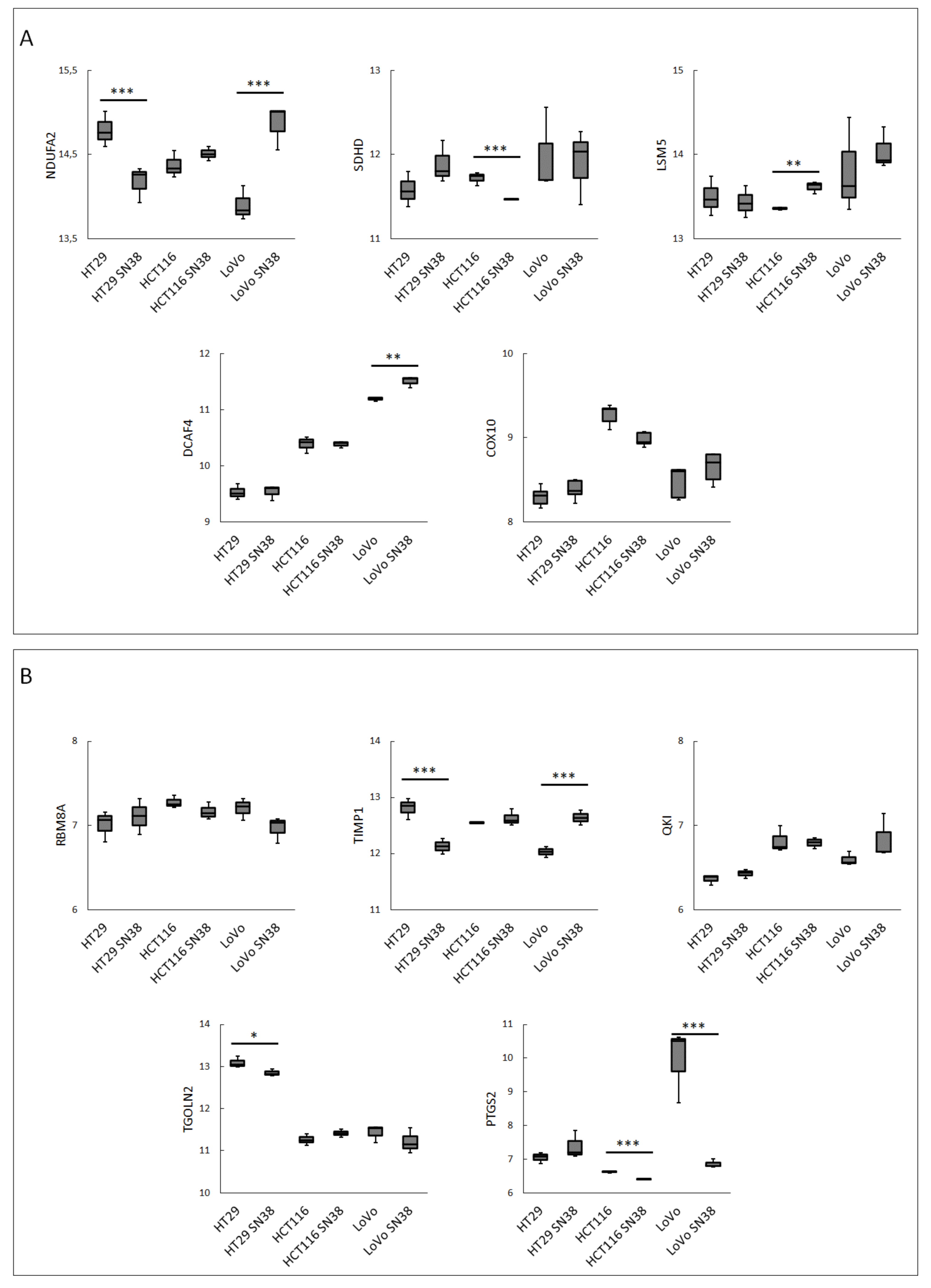
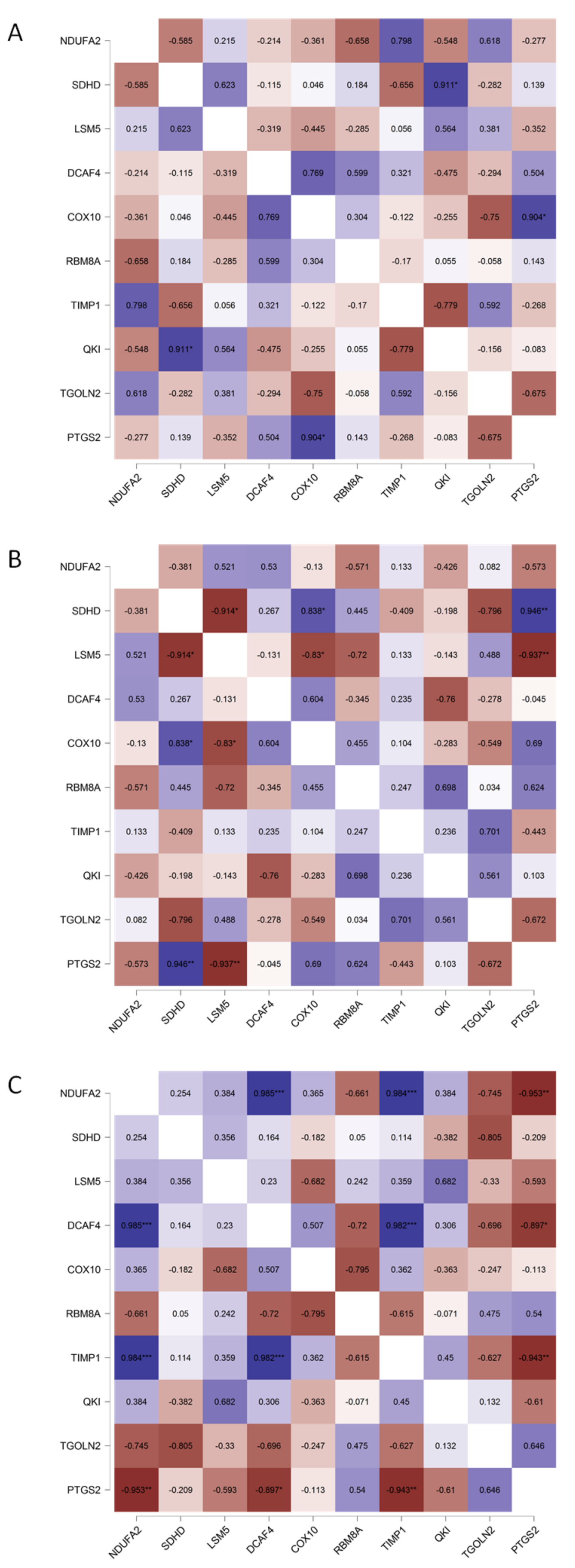
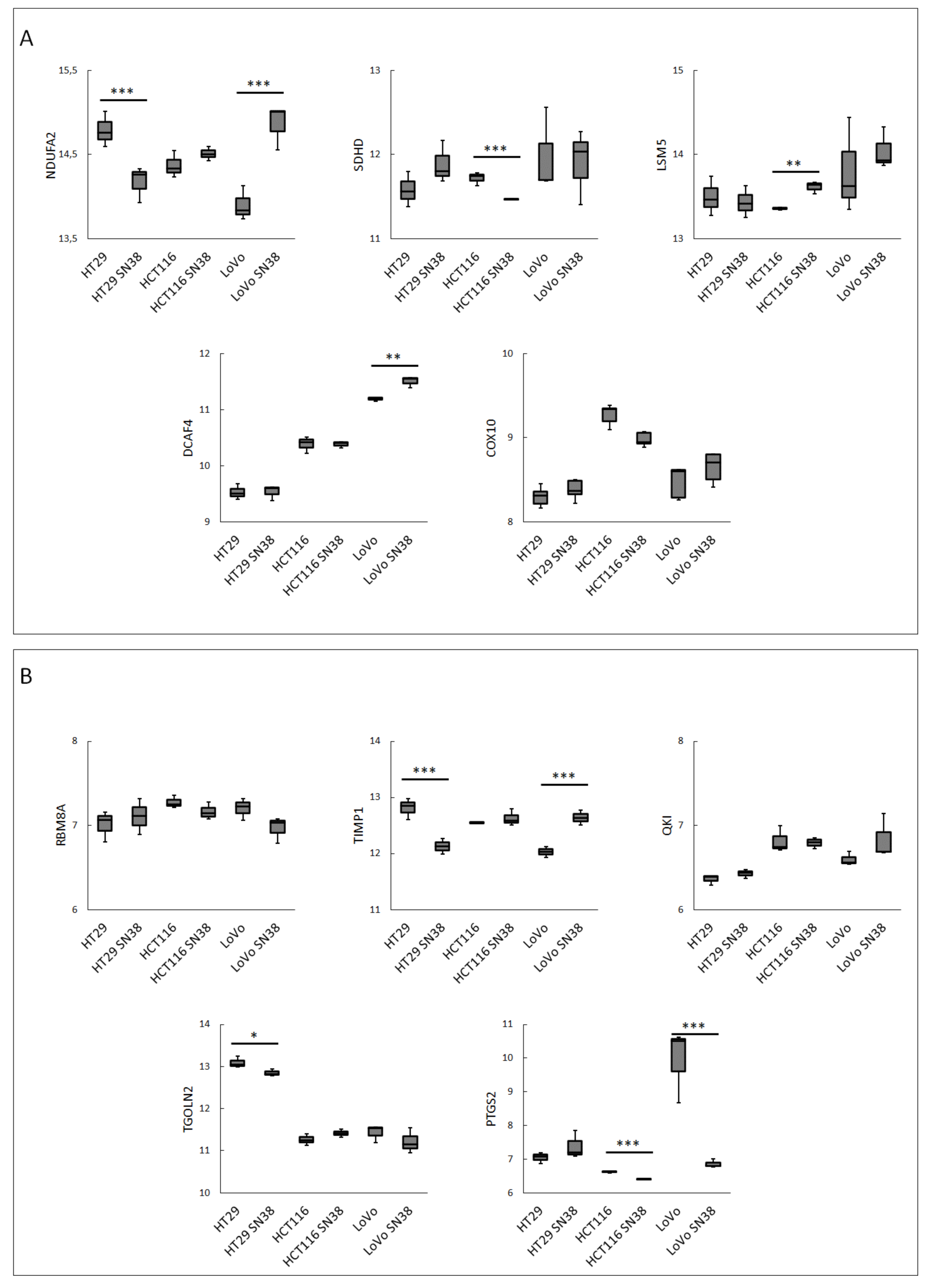
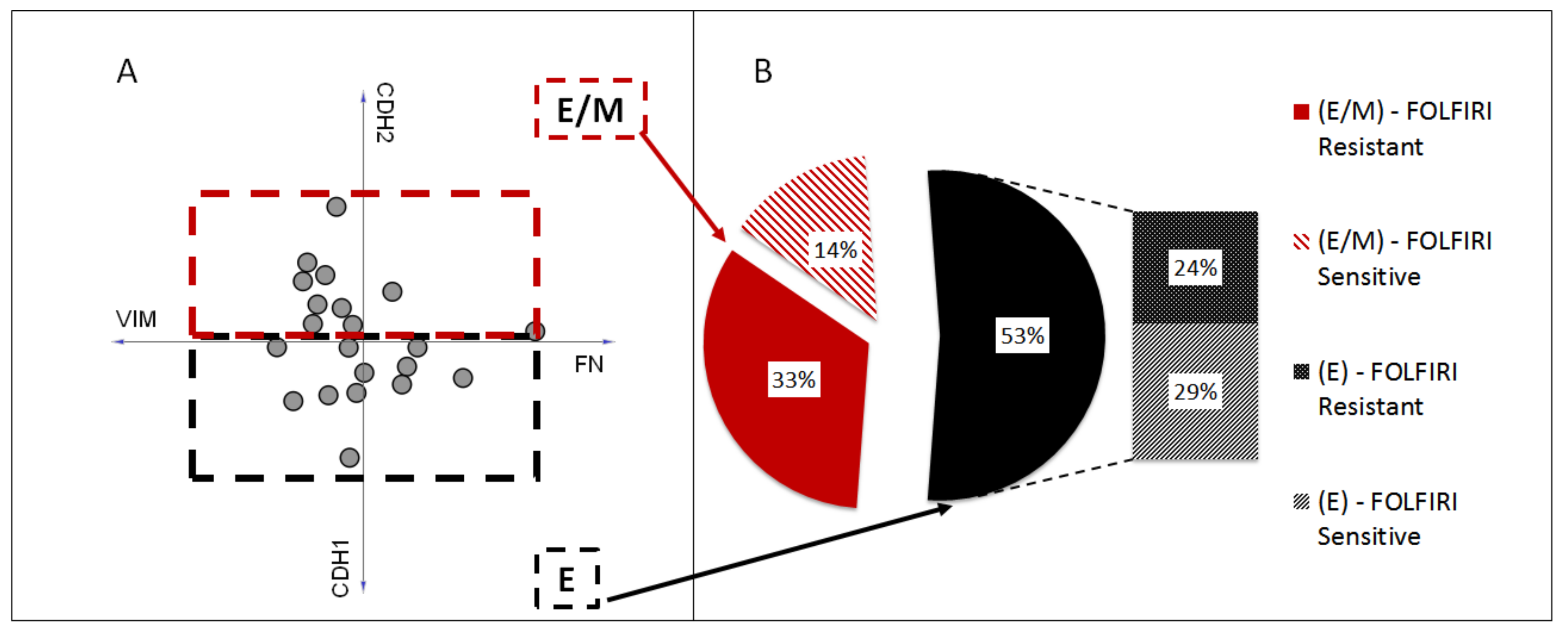
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-FU | 5 fluorouracil |
| ABC | ATP-binding cassette |
| ABCB1 | ATP-binding cassette subfamily B member 1 |
| ABCC1 | ATP-binding cassette subfamily C member 1 |
| ABCC2 | ATP-binding cassette subfamily C member 2 |
| ABCC4 | ATP-binding cassette subfamily C member 4 |
| ABCC5 | ATP-binding cassette subfamily C member 5 |
| ABCG2 | ATP-binding cassette subfamily G member 2 |
| ALDH | aldehyde dehydrogenase |
| APC | aminopentane carboxylic acid (7-ethyl-10[4-N-(5-aminopentanoicacid)-1-piperidino] carbonyloxycamptothecin) |
| CES1 | carboxylesterase 1 |
| CES2 | carboxylesterase 2 |
| CMS | consensus molecular subgroup |
| COX10 | cytochrome C Oxidase Assembly Homolog 10 |
| CPT-11 | camptothecin-11 |
| CYP3A4 | cytochrome P450 family 3 subfamily A member 4 |
| CYP3A5 | cytochrome P450 family 3 subfamily A member 5 |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| DCAF4 | DDB1 and CUL4-associated factor 4 |
| DEGs | differently expressed genes |
| FDR | false discovery rate |
| FN | fibronectin |
| FOLFIRI | chemotherapy combination scheme: folinic acid (leucovorin), fluorouracil, and irinotecan |
| FOLFOX | chemotherapy combination scheme: folinic acid (leucovorin), fluorouracil, and oxaliplatin |
| GEO | Gene Expression Omnibus |
| hBChE | butyrylcholinesterase |
| HIF-1 | hypoxia-inducible factor 1 |
| IL-17 | interleukin 17 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LCM | laser capture microdissection |
| LGR5 | leucine-rich repeat-containing G-protein-coupled receptor 5 |
| LSM5 | U6 small nuclear RNA and mRNA-degradation-associated protein 5 |
| MDR | multidrug resistance |
| NPC | (7-ethyl-10[4-amino-1-piperidino]carbonyloxycamptothecin) |
| NSCLC | non-small cell lung cancer |
| OATP | organic-anion-transporting polypeptides |
| OXPHOS | oxidative phosphorylation |
| RNA | ribonucleic acid |
| SN-38 | 7-ethyl-10-hydroxycamptothecin |
| TWIST-1 | Twist family BHLH transcription factor 1 |
| UGT1A1 | UDP glucuronosyltransferase family 1 member A1 |
| UGT1A10 | UDP glucuronosyltransferase family 1 member A10 |
| UGT1A7 | UDP glucuronosyltransferase family 1 member A7 |
| UGT1A9 | UDP glucuronosyltransferase family 1 member A9 |
| VEGF | vascular endothelial growth factor |
| VIM | vimentin |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D. ESMO Guidelines Working Group, Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii1–iii9. [Google Scholar] [CrossRef]
- Di Desidero, T.; Antonelli, A.; Orlandi, P.; Ferrari, S.M.; Fioravanti, A.; Alì, G.; Fontanini, G.; Basolo, F.; Francia, G.; Bocci, G. Synergistic efficacy of irinotecan and sunitinib combination in preclinical models of anaplastic thyroid cancer. Cancer Lett. 2017, 411, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tse-Dinh, Y.-C. Recent Advances in Use of Topoisomerase Inhibitors in Combination Cancer Therapy. Curr. Top. Med. Chem. 2019, 19, 730–740. [Google Scholar] [PubMed]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar]
- De Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Houghton, P.J.; Thompson, J.; Cheshire, P.J.; Hanna, S.K.; Richmond, L.B.; Lou, X.; Stewart, C.F. Altered irinotecan and SN-38 disposition after intravenous and oral administration of irinotecan in mice bearing human neuroblastoma xenografts. Clin. Cancer Res. 1998, 4, 455–462. [Google Scholar]
- Fujiwara, R.; Yoda, E.; Tukey, R.H. Species differences in drug glucuronidation: Humanized UDP-glucuronosyltransferase 1 mice and their application for predicting drug glucuronidation and drug-induced toxicity in humans. Drug Metab. Pharmacokinet. 2018, 33, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, L.; Harding, M.W.; Neubauer, M.; Langer, C.J.; Moore, M.; Ross, H.J.; Johnson, B.E.; Lynch, T.J. A phase II study of the safety and efficacy of the multidrug resistance inhibitor VX-710 combined with doxorubicin and vincristine in patients with recurrent small cell lung cancer. Cancer 2007, 109, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Slatter, J.G.; Schaaf, L.J.; Sams, J.P.; Feenstra, K.L.; Johnson, M.G.; Bombardt, P.A.; Cathcart, K.S.; Verburg, M.T.; Pearson, L.K.; Compton, L.D.; et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab. Dispos. 2000, 28, 423–433. [Google Scholar] [PubMed]
- Kryczka, J.; Boncela, J. Cell Migration Related to MDR-Another Impediment to Effective Chemotherapy? Molecules 2018, 23, 331. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.S.; Kawabe, T.; Ono, M.; Aoki, S.; Sumizawa, T.; Furukawa, T.; Uchiumi, T.; Wada, M.; Kuwano, M.; Akiyama, S.I. Effect of multidrug resistance-reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol. Pharmacol. 1999, 56, 1219–1228. [Google Scholar] [CrossRef]
- Paulík, A.; Nekvindová, J.; Filip, S. Irinotecan toxicity during treatment of metastatic colorectal cancer: Focus on pharmacogenomics and personalized medicine. Tumori 2020, 106, 87–94. [Google Scholar] [CrossRef]
- Kilic, E.; Spudich, A.; Kilic, U.; Rentsch, K.M.; Vig, R.; Matter, C.M.; Wunderli-Allenspach, H.; Fritschy, J.-M.; Bassetti, C.L.; Hermann, D.M. ABCC1: A gateway for pharmacological compounds to the ischaemic brain. Brain 2008, 131, 2679–2689. [Google Scholar] [CrossRef] [Green Version]
- Kryczka, J.; Sochacka, E.; Papiewska-Pająk, I.; Boncela, J. Implications of ABCC4–Mediated cAMP Eflux for CRC Migration. Cancers 2020, 12, 3547. [Google Scholar] [CrossRef]
- Ween, M.P.; Armstrong, M.A.; Oehler, M.K.; Ricciardelli, C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 220–256. [Google Scholar] [CrossRef]
- Petitprez, A.; Poindessous, V.; Ouaret, D.; Regairaz, M.; Bastian, G.; Guérin, E.; Escargueil, A.E.; Larsen, A.K. Acquired irinotecan resistance is accompanied by stable modifications of cell cycle dynamics independent of MSI status. Int. J. Oncol. 2013, 42, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.-X.; Yang, Y.; Zeng, L.; Patel, H.; Bo, L.; Lin, L.; Chen, Z.-S. Establishment and Characterization of an Irinotecan-Resistant Human Colon Cancer Cell Line. Front. Oncol. 2020, 10, 624954. [Google Scholar] [CrossRef]
- Kaur, G.; Dufour, J.M. Cell lines. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanta, P.; Ghosh, T.; Kaur, A.; Muthukumarappa, T. An innovative and cost-effective way to estimate alkaline phosphatase activity in in vitro cellular model systems. Int. J. Biochem. Mol. Biol. 2021, 12, 1–7. [Google Scholar] [PubMed]
- Pastor, D.M.; Poritz, L.S.; Olson, T.L.; Kline, C.L.; Harris, L.R.; Koltun, W.A.; Chinchilli, V.M.; Irby, R.B. Primary cell lines: False representation or model system? a comparison of four human colorectal tumors and their coordinately established cell lines. Int. J. Clin. Exp. Med. 2010, 3, 69–83. [Google Scholar] [PubMed]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.-T. The Role of Tumor Stroma in Cancer Progression and Prognosis: Emphasis on Carcinoma-Associated Fibroblasts and Non-small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Jensen, N.F.; Stenvang, J.; Beck, M.K.; Hanáková, B.; Belling, K.C.; Do, K.N.; Viuff, B.; Nygård, S.B.; Gupta, R.; Rasmussen, M.H.; et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol Oncol. 2015, 9, 1169–1185. [Google Scholar] [CrossRef]
- Rio, M.D.; Molina, F.; Bascoul-Mollevi, C.; Copois, V.; Bibeau, F.; Chalbos, P.; Bareil, C.; Kramar, A.; Salvetat, N.; Fraslon, C.; et al. Gene expression signature in advanced colorectal cancer patients select drugs and response for the use of leucovorin, fluorouracil, and irinotecan. J. Clin. Oncol. 2007, 25, 773. [Google Scholar] [CrossRef]
- Makondi, P.T.; Chu, C.-M.; Wei, P.-L.; Chang, Y.-J. Prediction of novel target genes and pathways involved in irinotecan-resistant colorectal cancer. PLoS ONE. 2017, 12, e0180616. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, M.; Yamashita, M.; Nishi, T.; Nakagawa, H. A Human ABC Transporter ABCC4 Gene SNP (rs11568658, 559 G > T, G187W) Reduces ABCC4-Dependent Drug Resistance. Cells 2019, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial-Mesenchymal Transition (EMT) with Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in Colorectal Cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol. Cell Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebio, A.; Lenz, H.-J. The Molecular Taxonomy of Colorectal Cancer: What’s New? Curr. Colorectal Cancer Rep. 2015, 11, 118–124. [Google Scholar] [CrossRef]
- Santamaría, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekulayev, V.; Mado, K.; Shevchuk, I.; Koit, A.; Kaldma, A.; Klepinin, A.; Timohhina, N.; Tepp, K.; Kandashvili, M.; Ounpuu, L.; et al. Metabolic remodeling in human colorectal cancer and surrounding tissues: Alterations in regulation of mitochondrial respiration and metabolic fluxes. Biochem. Biophys. Rep. 2015, 4, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Silva, V.S.e.; Chinen, L.T.D.; Abdallah, E.A.; Damascena, A.; Paludo, J.; Chojniak, R.; Dettino, A.L.A.; Mello, C.A.L.; Alves, V.S.; Fanelli, M.F. Early detection of poor outcome in patients with metastatic colorectal cancer: Tumor kinetics evaluated by circulating tumor cells. OncoTargets Ther. 2016, 9, 7503–7513. [Google Scholar] [CrossRef] [Green Version]
- Henry, J.T.; Johnson, B. Current and evolving biomarkers for precision oncology in the management of metastatic colorectal cancer. Chin. Clin. Oncol. 2019, 8, 49. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Ou, F.-S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of Consensus Molecular Subtype on Survival in Patients with Metastatic Colorectal Cancer: Results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- Przygodzka, P.; Papiewska-Pajak, I.; Bogusz, H.; Kryczka, J.; Sobierajska, K.; Kowalska, M.A.; Boncela, J. Neuromedin U is upregulated by Snail at early stages of EMT in HT29 colon cancer cells. Biochim. Biophys. Acta 2016, 1860, 2445–2453. [Google Scholar] [CrossRef]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Pasqualato, A.; Proietti, S.; Masiello, M.G.; Palombo, A.; Coluccia, P.; Canipari, R.; Catizone, A.; Ricci, G.; Harrath, A.H.; et al. Paradoxical E-cadherin increase in 5FU-resistant colon cancer is unaffected during mesenchymal-epithelial reversion induced by γ-secretase inhibition. Life Sci. 2016, 145, 174–183. [Google Scholar] [CrossRef]
- Jiang, Z.-S.; Sun, Y.-Z.; Wang, S.-M.; Ruan, J.-S. Epithelial-mesenchymal transition: Potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Ritvo, P.; Myers, R.E.; Serenity, M.; Gupta, S.; Inadomi, J.M.; Green, B.B.; Jerant, A.; Tinmouth, J.; Paszat, L.; Pirbaglou, M.; et al. Taxonomy for colorectal cancer screening promotion: Lessons from recent randomized controlled trials. Prev. Med. 2017, 101, 229–234. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Count in Network | FDR |
|---|---|---|
| Upregulated Genes | ||
| Oxidative phosphorylation | 48 of 131 | 7.97 × 10−54 |
| RNA degradation | 23 of 77 | 3.48 × 10−24 |
| Retrograde endocannabinoid signaling | 42 of 148 | 1.54 × 10−43 |
| Spliceosome | 34 of 130 | 3.52 × 10−34 |
| Thermogenesis | 50 of 228 | 3.39 × 10−47 |
| Citrate cycle (TCA cycle) | 4 of 30 | 0.0028 |
| Metabolic pathways | 52 of 1250 | 2.16 × 10−17 |
| Downregulated genes | ||
| Spliceosome | 60 of 130 | 4.04 × 10−72 |
| mRNA surveillance pathway | 21 of 89 | 4.61 × 10−19 |
| SNARE interactions in vesicular transport | 4 of 33 | 0.0044 |
| RNA transport | 19 of 159 | 1.75 × 10−12 |
| RNA degradation | 8 of 77 | 4.26 × 10−5 |
| Adipocytokine signaling pathway | 7 of 69 | 0.00018 |
| FoxO signaling pathway | 12 of 130 | 9.57 × 10−7 |
| AMPK signaling pathway | 10 of 120 | 1.89 × 10−5 |
| Glucagon signaling pathway | 7 of 100 | 0.0012 |
| Oxytocin signaling pathway | 10 of 149 | 8.76 × 10−5 |
| Insulin signaling pathway | 9 of 134 | 0.00021 |
| Apelin signaling pathway | 9 of 133 | 0.00021 |
| Tight junction | 11 of 167 | 4.24 × 10−5 |
| Endocrine resistance | 5 of 95 | 0.0221 |
| Signaling pathways regulating pluripotency of stem cells | 7 of 138 | 0.0059 |
| microRNAs in cancer | 7 of 149 | 0.0080 |
| mTOR signaling pathway | 7 of 148 | 0.0080 |
| Proteoglycans in cancer | 9 of 195 | 0.0024 |
| Rap1 signaling pathway | 9 of 203 | 0.0030 |
| Leukocyte transendothelial migration | 5 of 112 | 0.0401 |
| Focal adhesion | 8 of 197 | 0.0085 |
| Thermogenesis | 9 of 228 | 0.0059 |
| Herpes simplex infection | 7 of 181 | 0.0206 |
| PI3K-Akt signaling pathway | 10 of 348 | 0.0231 |
| Gene Name | Gene | Betweenness Centrality | Degree |
|---|---|---|---|
| NADH Ubiquinone Oxidoreductase Subunit A2 | NDUFA2 | 0.009082 | 48 |
| Succinate Dehydrogenase Complex Subunit D | SDHD | 0.013266 | 41 |
| LSM5 Homolog, U6 Small Nuclear RNA, and MRNA-Degradation-Associated | LSM5 | 7.02 × 10−4 | 36 |
| DDB1 and CUL4-Associated Factor 4 | DCAF4 | 0 | 25 |
| Cytochrome C Oxidase Assembly Factor Heme A: Farnesyltransferase | COX10 | 4.08 × 10−5 | 24 |
| RNA-Binding Motif Protein 8A | RBM8A | 0.007638 | 75 |
| TIMP Metallopeptidase Inhibitor 1 | TIMP1 | 0.031803 | 20 |
| KH Domain-Containing RNA-Binding | QKI | 0.035524 | 17 |
| Trans-Golgi Network Protein 2 | TGOLN2 | 0.062555 | 16 |
| Prostaglandin-Endoperoxide Synthase 2 | PTGS2 | 0.018124 | 14 |
| NDUFA2 | SDHD | LSM5 | DCAF4 | COX10 | RBM8A | TIMP1 | QKI | TGOLN2 | PTGS2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Arachidonic acid metabolism | x | |||||||||
| Biosynthesis of cofactors | x | |||||||||
| Biosynthesis of secondary metabolites | x | x | ||||||||
| Butanoate metabolism | x | |||||||||
| Carbon metabolism | x | |||||||||
| Chemical carcinogenesis | x | |||||||||
| Citrate cycle (TCA cycle) | x | |||||||||
| C-type lectin receptor signaling pathway | x | |||||||||
| HIF-1 signaling pathway | x | |||||||||
| Human cytomegalovirus infection | x | |||||||||
| Human papillomavirus infection | x | |||||||||
| IL-17 signaling pathway | x | |||||||||
| Metabolic pathways | x | x | x | x | ||||||
| Microbial metabolism in diverse environments | x | |||||||||
| mRNA surveillance pathway | x | |||||||||
| NF-kappa B signaling pathway | x | |||||||||
| Oxidative phosphorylation | x | x | x | |||||||
| Oxytocin signaling pathway | x | |||||||||
| Pathways in cancer | x | |||||||||
| Regulation of lipolysis in adipocytes | x | |||||||||
| Retrograde endocannabinoid signaling | x | x | ||||||||
| RNA degradation | x | |||||||||
| RNA transport | x | |||||||||
| Spliceosome | x | x | ||||||||
| Thermogenesis | x | x | x | |||||||
| TNF signaling pathway | x | |||||||||
| VEGF signaling pathway | x |
| Colon Adenocarcinoma | Rectum Adenocarcinoma | |||
|---|---|---|---|---|
| 5-Year Survival | ||||
| Gene | High | Low | High | Low |
| NDUFA2 | 72% | 58% | 19% | 67% |
| SDHD | 71% | 52% | 65% | 19% |
| LSM5 | 43% | 69% | 50% | 41% |
| DCAF4 | 61% | 71% | 61% | 45% |
| COX10 | 71% | 57% | 66% | 25% |
| RBM8A | 57% | 71% | 60% | 23% |
| TIMP1 | 53% | 69% | 34% | 89% |
| QKI | 64% | 63% | 57% | 0% |
| TGOLN2 | 64% | 63% | 62% | 47% |
| PTGS2 | 69% | 59% | 59% | 20% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryczka, J.; Boncela, J. Integrated Bioinformatics Analysis of the Hub Genes Involved in Irinotecan Resistance in Colorectal Cancer. Biomedicines 2022, 10, 1720. https://doi.org/10.3390/biomedicines10071720
Kryczka J, Boncela J. Integrated Bioinformatics Analysis of the Hub Genes Involved in Irinotecan Resistance in Colorectal Cancer. Biomedicines. 2022; 10(7):1720. https://doi.org/10.3390/biomedicines10071720
Chicago/Turabian StyleKryczka, Jakub, and Joanna Boncela. 2022. "Integrated Bioinformatics Analysis of the Hub Genes Involved in Irinotecan Resistance in Colorectal Cancer" Biomedicines 10, no. 7: 1720. https://doi.org/10.3390/biomedicines10071720
APA StyleKryczka, J., & Boncela, J. (2022). Integrated Bioinformatics Analysis of the Hub Genes Involved in Irinotecan Resistance in Colorectal Cancer. Biomedicines, 10(7), 1720. https://doi.org/10.3390/biomedicines10071720