
Venous Thromboembolism in Sepsis: From Bench to Bedside


Abstract
1. Introduction
2. Materials and Methods
3. From Bench
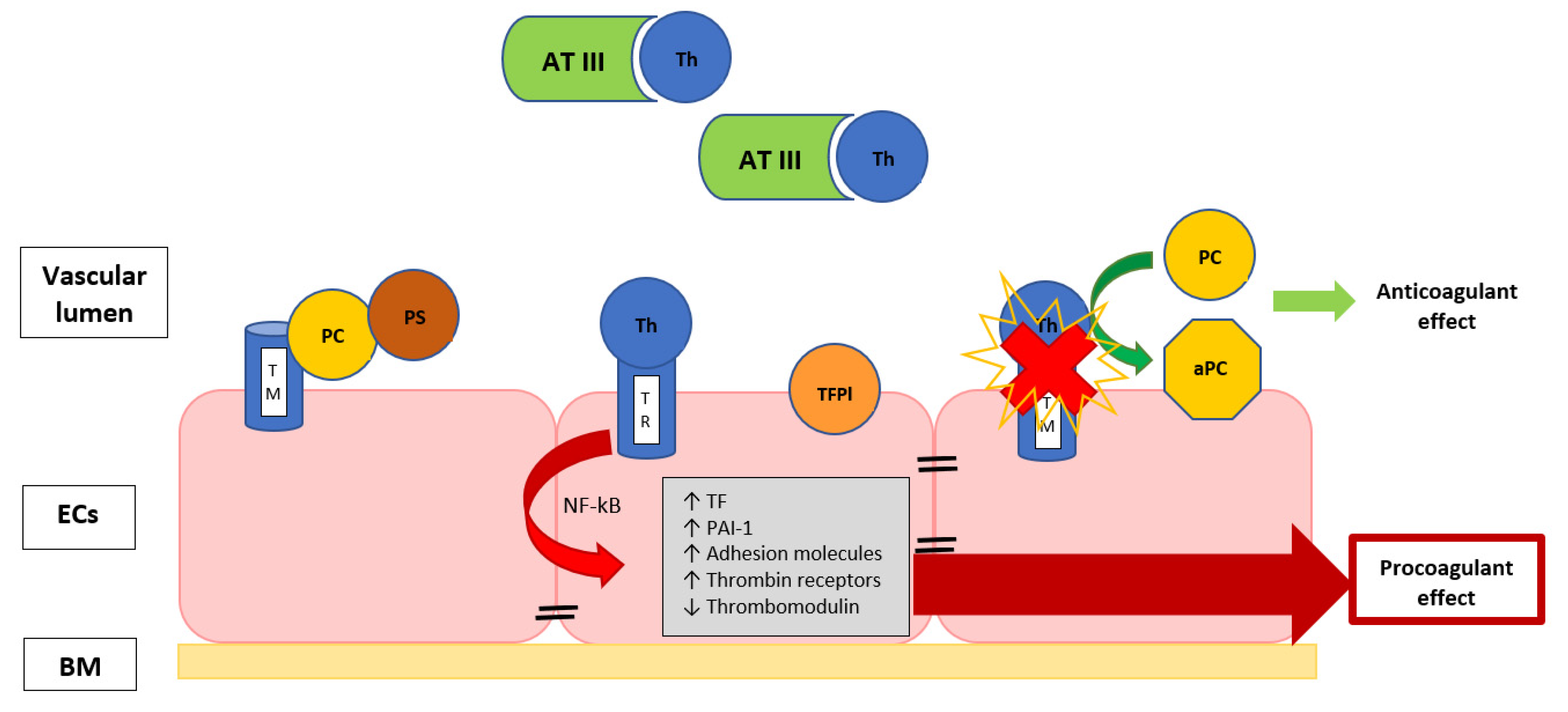
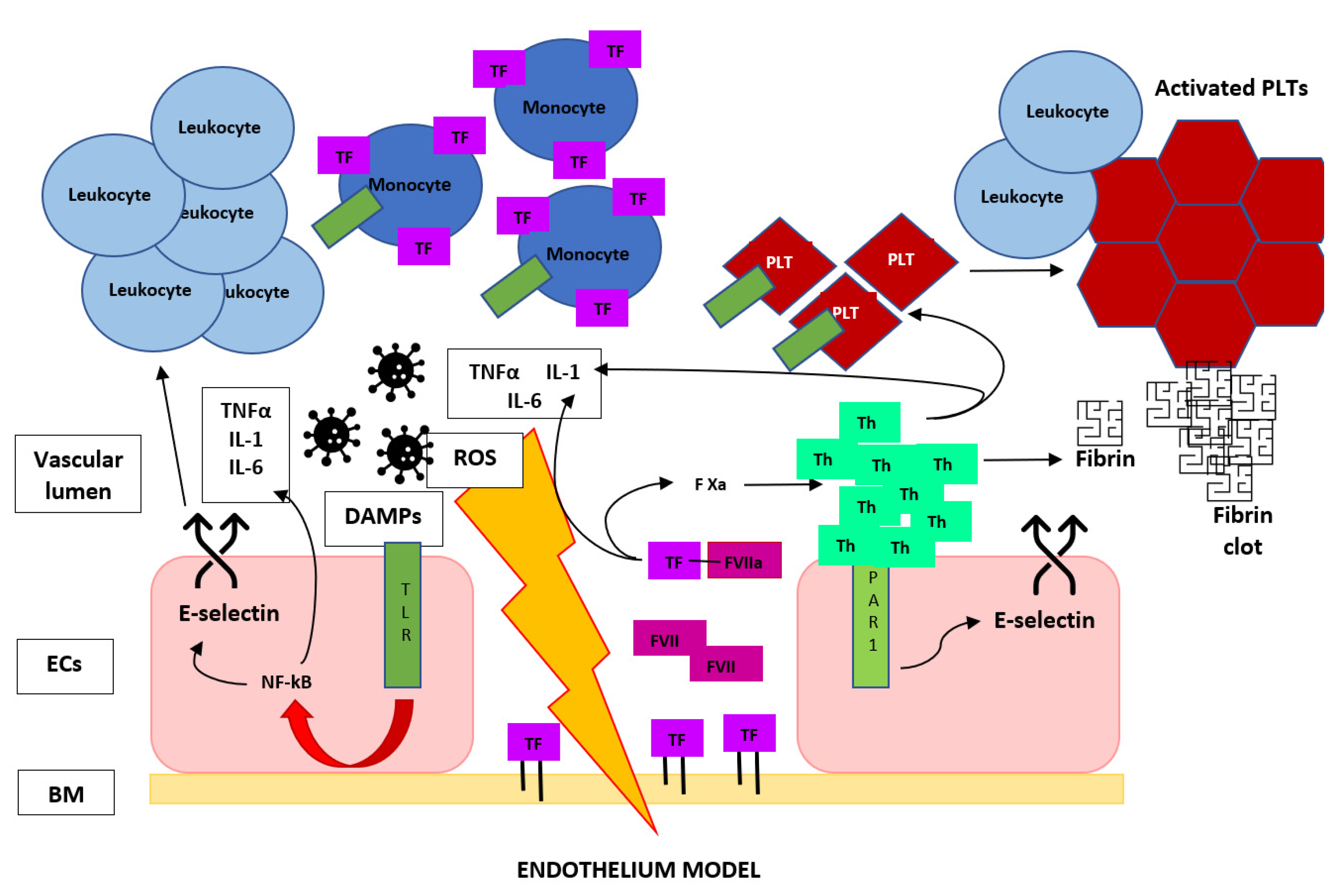
3.1. The Role of Endothelium
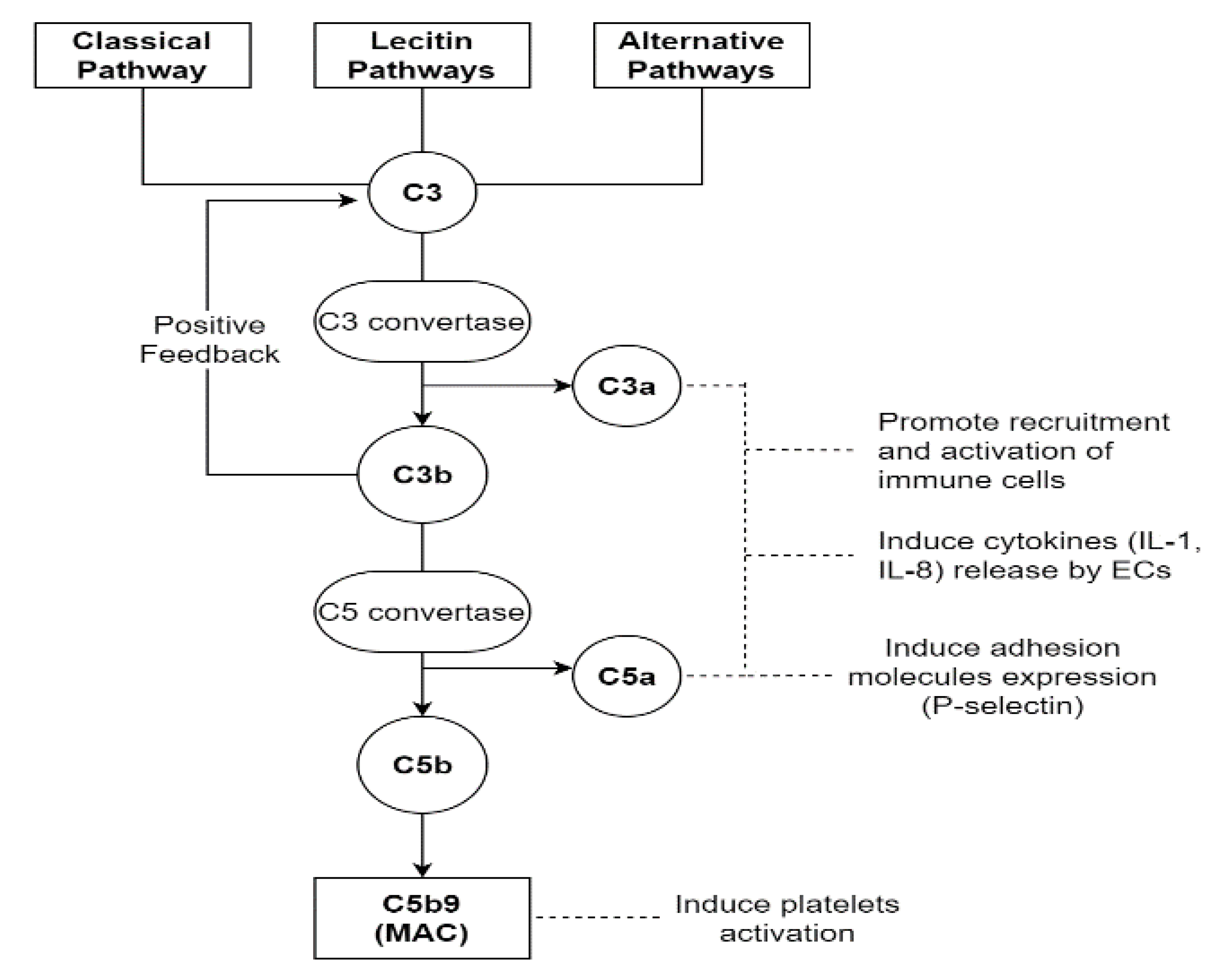
3.2. The Role of Coagulation Cascade
3.3. The Role of Leukocytes
3.4. COVID-19 Experience
4. To Bedside
4.1. Diagnosis Approach
4.2. Therapeutic Management
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ISTH. Steering Committee for World Thrombosis Day. Thrombosis: A major contributor to the global disease burden. J. Thromb. Haemost. JTH 2014, 12, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Donadini, M.P.; Ageno, W. Epidemiology and pathophysiology of venous thromboembolism: Similarities with atherothrombosis and the role of inflammation. Thromb. Haemost. 2015, 113, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Larizgoitia, I.; Audera-Lopez, C.; Prasopa-Plaizier, N.; Waters, H.; Bates, D.W. The global burden of unsafe medical care: Analytic modelling of observational studies. BMJ Qual. Saf. 2013, 22, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef]
- Liang, L.; Moore, B.; Soni, A. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2017: Statistical Brief #261. In Healthcare Cost and Utilization Project HCUP Statistical Briefs; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2020. [Google Scholar]
- Greco, E.; Lupia, E.; Bosco, O.; Vizio, B.; Montrucchio, G. Platelets and Multi-Organ Failure in Sepsis. Int. J. Mol. Sci. 2017, 18, 2200. [Google Scholar] [CrossRef]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med 2021, 47, 1181–1247. [Google Scholar] [CrossRef]
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Qu, M.; Nan, K.; Cao, H.; Cata, J.P.; Chen, W.; Miao, C. Review: The Emerging Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Associated Thrombosis. Front. Cell Infect. Microbiol. 2021, 11, 653228. [Google Scholar] [CrossRef]
- Yang, S.; Qi, H.; Kan, K.; Chen, J.; Xie, H.; Guo, X.; Zhang, L. Neutrophil Extracellular Traps Promote Hypercoagulability in Patients With Sepsis. Shock Augusta Ga 2017, 47, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Garza, E.; Jerjes-Sanchez, C.; Navarrete, A.; Joya-Harrison, J.; Rodriguez, D. Venous thromboembolism: Thrombosis, inflammation, and immunothrombosis for clinicians. J. Thromb. Thrombolysis 2017, 44, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Tritschler, T.; Kahn, S.R.; Rodger, M.A. Venous thromboembolism. Lancet 2021, 398, 64–77. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Hafizi, S.; Rezaei, N. Inflammation in venous thromboembolism: Cause or consequence? Int. Immunopharmacol. 2015, 28, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Tanguay, J.-F.; Geoffroy, P.; Sirois, M.G.; Libersan, D.; Kumar, A.; Schaub, R.G.; Merhi, Y. Prevention of in-stent restenosis via reduction of thrombo-inflammatory reactions with recombinant P-selectin glycoprotein ligand-1. Thromb. Haemost. 2014, 91, 1186–1193. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Chang, J.C. Sepsis and septic shock: Endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb. J. 2019, 17, 10. [Google Scholar] [CrossRef]
- Chang, J.C. Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”. Life 2022, 12, 220. [Google Scholar] [CrossRef]
- Opal, S.M. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit. Care Med. 2020, 28, S77–S80. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Levin, E.G.; Santell, L.; Osborn, K.G. The expression of endothelial tissue plasminogen activator in vivo: A function defined by vessel size and anatomic location. J. Cell Sci. 1997, 110 (Suppl. 2), 139–148. [Google Scholar] [CrossRef]
- Takahashi, K.; Uwabe, Y.; Sawasaki, Y.; Kiguchi, T.; Nakamura, H.; Kashiwabara, K.; Yagyu, H.; Matsuoka, T. Increased secretion of urokinase-type plasminogen activator by human lung microvascular endothelial cells. Am. J. Physiol. 1998, 275, L47–L54. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Maury, E.; Lehoux, S.; Guidet, B.; Offenstadt, G. The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010, 36, 1286–1298. [Google Scholar] [CrossRef]
- Vallet, B. Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: A role in organ dysfunction? Crit. Care 2003, 7, 130–138. [Google Scholar] [CrossRef]
- McCormack, J.J.; da Silva, M.L.; Ferraro, F.; Patella, F.; Cutler, D.F. Weibel-Palade bodies at a glance. J. Cell Sci. 2017, 130, 3611–3617. [Google Scholar] [CrossRef]
- Barrionuevo, N.; Gatica, S.; Olivares, P.; Cabello-Verrugio, C.; Simon, F. Endothelial Cells Exhibit Two Waves of P-selectin Surface Aggregation Under Endotoxic and Oxidative Conditions. Protein J. 2019, 38, 667–674. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Köllnberger, M.; Wakefield, T.W.; Lämmle, B.; Massberg, S.; Wagner, D.D. Von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef]
- Petri, B.; Broermann, A.; Li, H.; Khandoga, A.G.; Zarbock, A.; Krombach, F.; Goerge, T.; Schneider, S.W.; Jones, C.; Nieswandt, B.; et al. von Willebrand factor promotes leukocyte extravasation. Blood 2010, 116, 4712–4719. [Google Scholar] [CrossRef]
- Kuebler, W.M.; Borges, J.; Sckell, A.; Kuhnle, G.E.; Bergh, K.; Messmer, K.; Goetz, A.E. Role of L-selectin in leukocyte sequestration in lung capillaries in a rabbit model of endotoxemia. Am. J. Respir. Crit. Care Med. 2000, 161, 36–43. [Google Scholar] [CrossRef]
- Matsukawa, A.; Lukacs, N.W.; Hogaboam, C.M.; Knibbs, R.N.; Bullard, D.C.; Kunkel, S.L.; Stoolman, L.M. Mice genetically lacking endothelial selectins are resistant to the lethality in septic peritonitis. Exp. Mol. Pathol. 2002, 72, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Gardinali, M.; Borrelli, E.; Chiara, O.; Lundberg, C.; Padalino, P.; Conciato, L.; Cafaro, C.; Lazzi, S.; Luzi, P.; Giomarelli, P.P.; et al. Inhibition of CD11-CD18 complex prevents acute lung injury and reduces mortality after peritonitis in rabbits. Am. J. Respir. Crit. Care Med. 2000, 161, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.; Pittet, J.-F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 2015, 28, 227–236. [Google Scholar] [CrossRef]
- Reidy, M.A.; Schwartz, S.M. Endothelial injury and regeneration. IV. Endotoxin: A nondenuding injury to aortic endothelium. Lab. Investig. J. Tech. Methods Pathol. 1983, 48, 25–34. [Google Scholar]
- Reidy, M.A.; Bowyer, D.E. Scanning electron microscopy: Morphology of aortic endothelium following injury by endotoxin and during subsequent repair. Atherosclerosis 1977, 26, 319–328. [Google Scholar] [CrossRef]
- Leclerc, J.; Pu, Q.; Corseaux, D.; Haddad, E.; Decoene, C.; Bordet, R.; Six, I.; Jude, B.; Vallet, B. A single endotoxin injection in the rabbit causes prolonged blood vessel dysfunction and a procoagulant state. Crit. Care Med. 2000, 28, 3672–3678. [Google Scholar] [CrossRef]
- Wang, P.; Ba, Z.F.; Chaudry, I.H. Endothelium-dependent relaxation is depressed at the macro- and microcirculatory levels during sepsis. Am. J. Physiol. 1995, 269, R988–R994. [Google Scholar] [CrossRef]
- Wylam, M.E.; Samsel, R.W.; Umans, J.G.; Mitchell, R.W.; Leff, A.R.; Schumacker, P.T. Endotoxin in vivo impairs endothelium-dependent relaxation of canine arteries in vitro. Am. Rev. Respir. Dis. 1990, 142, 1263–1267. [Google Scholar] [CrossRef]
- McKenna, T.M.; Martin, F.M.; Chernow, B.; Briglia, F.A. Vascular endothelium contributes to decreased aortic contractility in experimental sepsis. Circ. Shock. 1986, 19, 267–273. [Google Scholar]
- Lee, M.M.; Schuessler, G.B.; Chien, S. Time-dependent effects of endotoxin on the ultrastructure of aortic endothelium. Artery 1988, 15, 71–89. [Google Scholar]
- Brooks, E.G.; Trotman, W.; Wadsworth, M.P.; Taatjes, D.J.; Evans, M.F.; Ittleman, F.P.; Callas, P.W.; Esmon, C.T.; Bovill, E.G. Valves of the deep venous system: An overlooked risk factor. Blood 2009, 114, 1276–1279. [Google Scholar] [CrossRef][Green Version]
- Welsh, J.D.; Hoofnagle, M.H.; Bamezai, S.; Oxendine, M.; Lim, L.; Hall, J.D.; Yang, J.; Schultz, S.; Engel, J.D.; Kume, T.; et al. Hemodynamic regulation of perivalvular endothelial gene expression prevents deep venous thrombosis. J. Clin. Investig. 2019, 129, 5489–5500. [Google Scholar] [CrossRef]
- Chang, J.C. Thrombogenesis and thrombotic disorders based on “two-path unifying theory of hemostasis”: Philosophical, physiological, and phenotypical interpretation. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2018, 29, 585–595. [Google Scholar] [CrossRef]
- Chang, J.C. Hemostasis based on a novel “two-path unifying theory” and classification of hemostatic disorders. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2018, 29, 573–584. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef]
- Wallis, R.; Mitchell, D.A.; Schmid, R.; Schwaeble, W.J.; Keeble, A.H. Paths reunited: Initiation of the classical and lectin pathways of complement activation. Immunobiology 2010, 215, 1–11. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Trieu, K.; Sikora, L.; Sriramarao, P.; DiScipio, R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J. Immunol. 2002, 169, 2102–2110. [Google Scholar] [CrossRef]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef]
- Morgan, B.P. The membrane attack complex as an inflammatory trigger. Immunobiology 2016, 221, 747–751. [Google Scholar] [CrossRef]
- Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Investig. 2012, 122, 2331–2336. [Google Scholar] [CrossRef]
- Pawlinski, R.; Mackman, N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb. Res. 2010, 125 (Suppl. 1), S70–S73. [Google Scholar] [CrossRef]
- Morrissey, J.H. Tissue factor interactions with factor VII: Measurement and clinical significance of factor VIIa in plasma. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 1995, 6 (Suppl. 1), S14–S19. [Google Scholar] [CrossRef]
- Camerer, E.; Huang, W.; Coughlin, S.R. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. USA 2000, 97, 5255–5260. [Google Scholar] [CrossRef] [PubMed]
- Levi, M. The coagulant response in sepsis and inflammation. Hamostaseologie 2010, 30, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef]
- Biemond, B.J.; Levi, M.; ten Cate, H.; Soule, H.R.; Morris, L.D.; Foster, D.L.; Bogowitz, C.A.; van der Poll, T.; Büller, H.R.; ten Cate, J.W. Complete inhibition of endotoxin-induced coagulation activation in chimpanzees with a monoclonal Fab fragment against factor VII/VIIa. Thromb. Haemost. 1995, 73, 223–230. [Google Scholar] [CrossRef]
- Sharma, L.; Melis, E.; Hickey, M.J.; Clyne, C.D.; Erlich, J.; Khachigian, L.M.; Davenport, P.; Morand, E.; Carmeliet, P.; Tipping, P.G. The cytoplasmic domain of tissue factor contributes to leukocyte recruitment and death in endotoxemia. Am. J. Pathol. 2004, 165, 331–340. [Google Scholar] [CrossRef]
- Van Deventer, S.J.; Büller, H.R.; ten Cate, J.W.; Aarden, L.A.; Hack, C.E.; Sturk, A. Experimental endotoxemia in humans: Analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood 1990, 76, 2520–2526. [Google Scholar] [CrossRef]
- Van der Poll, T.; Büller, H.R.; ten Cate, H.; Wortel, C.H.; Bauer, K.A.; van Deventer, S.J.; Hack, C.E.; Sauerwein, H.P.; Rosenberg, R.D.; ten Cate, J.W. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med. 1990, 322, 1622–1627. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. Pathologies at the nexus of blood coagulation and inflammation: Thrombin in hemostasis, cancer, and beyond. J. Mol. Med. 2013, 91, 1257–1271. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949. [Google Scholar] [CrossRef]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Investig. 1999, 103, 879–887. [Google Scholar] [CrossRef]
- Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M.; Stafforini, D.M. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit. Care Med. 2002, 30, S294–S301. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Mosad, E.; Elsayh, K.I.; Eltayeb, A.A. Tissue factor pathway inhibitor and P-selectin as markers of sepsis-induced non-overt disseminated intravascular coagulopathy. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2011, 17, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [PubMed]
- Seeley, E.J.; Matthay, M.A.; Wolters, P.J. Inflection points in sepsis biology: From local defense to systemic organ injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L355–L363. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Chi, H.; Sun, L. Neutrophils of Scophthalmus maximus produce extracellular traps that capture bacteria and inhibit bacterial infection. Dev. Comp. Immunol. 2016, 56, 7–12. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- De Buhr, N.; Neumann, A.; Jerjomiceva, N.; von Köckritz-Blickwede, M.; Baums, C.G. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology 2014, 160, 385–395. [Google Scholar] [CrossRef]
- Laarman, A.J.; Mijnheer, G.; Mootz, J.M.; van Rooijen, W.J.; Ruyken, M.; Malone, C.L.; Heezius, E.C.; Ward, R.; Milligan, G.; van Strijp, J.A.; et al. Staphylococcus aureus Staphopain A inhibits CXCR2-dependent neutrophil activation and chemotaxis. EMBO J. 2012, 31, 3607–3619. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef]
- Yago, T.; Liu, Z.; Ahamed, J.; McEver, R.P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 2018, 132, 1426–1437. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.-B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Diaz, J.A.; Fuchs, T.A.; Jackson, T.O.; Hovinga, J.A.K.; Lämmle, B.; Henke, P.K.; Myers, D.D.; Wagner, D.D.; Wakefield, T.W. For the Michigan Research Venous Group* (2013) Plasma DNA is Elevated in Patients with Deep Vein Thrombosis. J. Vasc. Surg. Venous Lymphat Disord. 2013, 1, 341–348. [Google Scholar] [CrossRef]
- Van Montfoort, M.L.; Stephan, F.; Lauw, M.N.; Hutten, B.A.; Van Mierlo, G.J.; Solati, S.; Middeldorp, S.; Meijers, J.C.M.; Zeerleder, S. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 147–151. [Google Scholar] [CrossRef]
- Granger, V.; Faille, D.; Marani, V.; Noël, B.; Gallais, Y.; Szely, N.; Flament, H.; Pallardy, M.; Chollet-Martin, S.; de Chaisemartin, L. Human blood monocytes are able to form extracellular traps. J. Leukoc. Biol. 2017, 102, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, J.M.; Gordon, A.E.; Henke, P.K. Resolution of Deep Venous Thrombosis: Proposed Immune Paradigms. Int. J. Mol. Sci. 2020, 21, 2080. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.E.; De Luca, M.; McNally, T.; Michelson, A.D.; Andrews, R.K.; Berndt, M.C. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood 2001, 98, 1440–1447. [Google Scholar] [CrossRef]
- Das, R.; Burke, T.; Plow, E.F. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 2007, 110, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Eagleton, M.J.; Henke, P.K.; Luke, C.E.; Hawley, A.E.; Bedi, A.; Knipp, B.S.; Wakefield, T.W.; Greenfield, L.J. Southern Association for Vascular Surgery William J. von Leibig Award. Inflammation and intimal hyperplasia associated with experimental pulmonary embolism. J. Vasc. Surg. 2002, 36, 581–588. [Google Scholar] [CrossRef]
- Varma, M.R.; Varga, A.J.; Knipp, B.S.; Sukheepod, P.; Upchurch, G.R.; Kunkel, S.L.; Wakefield, T.W.; Henke, P.K. Neutropenia impairs venous thrombosis resolution in the rat. J. Vasc. Surg. 2003, 38, 1090–1098. [Google Scholar] [CrossRef]
- Colling, M.E.; Tourdot, B.E.; Kanthi, Y. Inflammation, Infection and Venous Thromboembolism. Circ. Res. 2021, 128, 2017–2036. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453. [Google Scholar] [CrossRef]
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80. [Google Scholar] [CrossRef]
- Pieralli, F.; Pomero, F.; Giampieri, M.; Marcucci, R.; Prisco, D.; Luise, F.; Mancini, A.; Milia, A.; Sammicheli, L.; Tassinari, I.; et al. Incidence of deep vein thrombosis through an ultrasound surveillance protocol in patients with COVID-19 pneumonia in non-ICU setting: A multicenter prospective study. PLoS ONE 2021, 16, e0251966. [Google Scholar] [CrossRef]
- Kollias, A.; Kyriakoulis, K.G.; Lagou, S.; Kontopantelis, E.; Stergiou, G.S.; Syrigos, K. Venous thromboembolism in COVID-19: A systematic review and meta-analysis. Vasc. Med. 2021, 26, 415–425. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Liu, W.; Obana, T.; Sata, M. Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 12081. [Google Scholar] [CrossRef]
- Papadakis, D.D.; Politou, M.; Kompoti, M.; Vagionas, D.; Kostakou, E.; Theodoulou, D.; Kaniaris, E.; Rovina, N.; Panayiotakopoulos, G.; Dimopoulos, S.; et al. Immunostimulation and Coagulopathy in COVID-19 Compared to Patients With H1N1 Pneumonia or Bacterial Sepsis. Vivo Athens Greece 2022, 36, 954–960. [Google Scholar] [CrossRef]
- Hariri, L.P.; North, C.M.; Shih, A.R.; Israel, R.A.; Maley, J.H.; Villalba, J.A.; Vinarsky, V.; Rubin, J.; Okin, D.A.; Sclafani, A.; et al. Lung Histopathology in Coronavirus Disease 2019 as Compared With Severe Acute Respiratory Sydrome and H1N1 Influenza: A Systematic Review. Chest 2021, 159, 73–84. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X.-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.-T. SARS-CoV-2 Mediated Endothelial Dysfunction: The Potential Role of Chronic Oxidative Stress. Front. Physiol. 2020, 11, 605908. [Google Scholar] [CrossRef]
- Ali, M.A.M.; Spinler, S.A. COVID-19 and thrombosis: From bench to bedside. Trends Cardiovasc. Med. 2021, 31, 143–160. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.; Righy, C.; Franco, S.; Souza, T.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Zhu, W.; Zheng, Y.; Yu, M.; Wei, J.; Zhang, Y.; Topchyan, P.; Nguyen, C.; Janecke, R.; Kreuziger, L.B.; White, G.C.; et al. SARS-CoV-2 receptor binding domain-specific antibodies activate platelets with features resembling the pathogenic antibodies in heparin-induced thrombocytopenia. Res. Sq. 2021, preprint. [Google Scholar]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Palankar, R.; Wesche, J.; Handtke, S.; Wolff, M.; Aurich, K.; Lalk, M.; Methling, K.; Völker, U.; et al. Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia. Blood 2021, 138, 2256–2268. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Scientific and Standardization Committee on DIC, and the Scientific and Standardization Committee on Perioperative and Critical Care of the International Society on Thrombosis and Haemostasis Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. JTH 2019, 17, 1989–1994. [Google Scholar]
- Luo, C.; Hu, H.; Gong, J.; Zhou, Y.; Chen, Z.; Cai, S. The Value of Thromboelastography in the Diagnosis of Sepsis-Induced Coagulopathy. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2020, 26, 1076029620951847. [Google Scholar] [CrossRef]
- Müller, M.C.; Meijers, J.C.M.; Vroom, M.B.; Juffermans, N.P. Utility of thromboelastography and/or thromboelastometry in adults with sepsis: A systematic review. Crit. Care 2014, 18, R30. [Google Scholar] [CrossRef] [PubMed]
- Ramacciotti, E.; Blackburn, S.; Hawley, A.E.; Vandy, F.; Ballard-Lipka, N.; Stabler, C.; Baker, N.; Guire, K.E.; Rectenwald, J.E.; Henke, P.K.; et al. Evaluation of soluble P-selectin as a marker for the diagnosis of deep venous thrombosis. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2011, 17, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Pomero, F.; Dentali, F.; Borretta, V.; Bonzini, M.; Melchio, R.; Douketis, J.D.; Fenoglio, L.M. Accuracy of emergency physician-performed ultrasonography in the diagnosis of deep-vein thrombosis: A systematic review and meta-analysis. Thromb. Haemost. 2013, 109, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.V.; Meyer, G. The 2019 ESC Guidelines on the Diagnosis and Management of Acute Pulmonary Embolism. Eur. Heart J. 2019, 40, 3453–3455. [Google Scholar] [CrossRef]
- Squizzato, A.; Pomero, F.; Allione, A.; Priotto, R.; Riva, N.; Huisman, M.V.; Klok, F.A.; Stein, P.D.; Guasti, L.; Fenoglio, L.; et al. Diagnostic accuracy of magnetic resonance imaging in patients with suspected pulmonary embolism: A bivariate meta-analysis. Thromb. Res. 2017, 154, 64–72. [Google Scholar] [CrossRef]
- Vardi, M.; Ghanem-Zoubi, N.O.; Zidan, R.; Yurin, V.; Bitterman, H. Venous thromboembolism and the utility of the Padua Prediction Score in patients with sepsis admitted to internal medicine departments. J. Thromb. Haemost. JTH 2013, 11, 467–473. [Google Scholar] [CrossRef]
- Gibson, C.M.; Spyropoulos, A.C.; Cohen, A.T.; Hull, R.D.; Goldhaber, S.Z.; Yusen, R.D.; Hernandez, A.F.; Korjian, S.; Daaboul, Y.; Gold, A.; et al. The IMPROVEDD VTE Risk Score: Incorporation of D-Dimer into the IMPROVE Score to Improve Venous Thromboembolism Risk Stratification. TH Open Companion J. Thromb. Haemost. 2017, 1, e56–e65. [Google Scholar] [CrossRef]
- Rosenberg, D.; Eichorn, A.; Alarcon, M.; McCullagh, L.; McGinn, T.; Spyropoulos, A.C. External validation of the risk assessment model of the International Medical Prevention Registry on Venous Thromboembolism (IMPROVE) for medical patients in a tertiary health system. J. Am. Heart. Assoc. 2014, 3, e001152. [Google Scholar] [CrossRef]
- Goldin, M.; Lin, S.K.; Kohn, N.; Qiu, M.; Cohen, S.L.; Barish, M.A.; Gianos, E.; Diaz, A.; Richardson, S.; Giannis, D.; et al. External validation of the IMPROVE-DD risk assessment model for venous thromboembolism among inpatients with COVID-19. J. Thromb. Thrombolysis 2021, 52, 1032–1035. [Google Scholar] [CrossRef]
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.D.; Wang, G.; Toh, C.-H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2019, 200, 869–880. [Google Scholar] [CrossRef]
- Bakirci, E.M.; Topcu, S.; Kalkan, K.; Tanboga, I.H.; Borekci, A.; Sevimli, S.; Acikel, M. The role of the nonspecific inflammatory markers in determining the anatomic extent of venous thromboembolism. Clin. Appl. Thromb. Off. J. Int. Acad. Clin. Appl. Thromb. 2015, 21, 181–185. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Stafford, A.R.; Dwivedi, D.J.; Kim, P.Y.; Fox-Robichaud, A.E.; Weitz, J.I.; Liaw, P.C. Cell-Free DNA Modulates Clot Structure and Impairs Fibrinolysis in Sepsis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2544–2553. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Toltl, L.J.; Swystun, L.L.; Pogue, J.; Liaw, K.-L.; Weitz, J.I.; Cook, D.J.; Fox-Robichaud, A.E.; Liaw, P.C.; Canadian Critical Care Translational Biology Group. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit. Care 2012, 16, R151. [Google Scholar] [CrossRef]
- Rhodes, A.; Wort, S.J.; Thomas, H.; Collinson, P.; Bennett, E.D. Plasma DNA concentration as a predictor of mortality and sepsis in critically ill patients. Crit. Care 2006, 10, R60. [Google Scholar] [CrossRef]
- Prével, R.; Dupont, A.; Labrouche-Colomer, S.; Garcia, G.; Dewitte, A.; Rauch, A.; Goutay, J.; Caplan, M.; Jozefowicz, E.; Lanoix, J.P.; et al. Plasma Markers of Neutrophil Extracellular Trap Are Linked to Survival but Not to Pulmonary Embolism in COVID-19-Related ARDS Patients. Front. Immunol. 2012, 13, 851497. [Google Scholar] [CrossRef]
- Grässle, S.; Huck, V.; Pappelbaum, K.I.; Gorzelanny, C.; Aponte-Santamaría, C.; Baldauf, C.; Gräter, F.; Schneppenheim, R.; Obser, T.; Schneider, S.W. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1382–1389. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, C.; Zhang, X.; Wang, S.; Zhu, R.; Zhou, A.; Chen, S.; Feng, J. Heparin improves alveolarization and vascular development in hyperoxia-induced bronchopulmonary dysplasia by inhibiting neutrophil extracellular traps. Biochem. Biophys. Res. Commun. 2020, 522, 33–39. [Google Scholar] [CrossRef]
- Longstaff, C.; Hogwood, J.; Gray, E.; Komorowicz, E.; Varjú, I.; Varga, Z.; Kolev, K. Neutralisation of the anti-coagulant effects of heparin by histones in blood plasma and purified systems. Thromb. Haemost. 2016, 115, 591–599. [Google Scholar]
- Pouplard, C.; May, M.A.; Iochmann, S.; Amiral, J.; Vissac, A.M.; Marchand, M.; Gruel, Y. Antibodies to platelet factor 4-heparin after cardiopulmonary bypass in patients anticoagulated with unfractionated heparin or a low-molecular-weight heparin: Clinical implications for heparin-induced thrombocytopenia. Circulation 1999, 99, 2530–2536. [Google Scholar] [CrossRef]
- Lelliott, P.M.; Momota, M.; Shibahara, T.; Lee, M.S.J.; Smith, N.I.; Ishii, K.J.; Coban, C. Heparin induces neutrophil elastase-dependent vital and lytic NET formation. Int. Immunol. 2020, 32, 359–368. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Levy, M.; Williams, M.D.; Douglas, I.; Artigas, A.; Antonelli, M.; Wyncoll, D.; Janes, J.; Booth, F.V.; Wang, D.; et al. Prophylactic heparin in patients with severe sepsis treated with drotrecogin alfa (activated). Am. J. Respir. Crit. Care Med. 2007, 176, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Crowther, M.; Meade, M.; Rabbat, C.; Griffith, L.; Schiff, D.; Geerts, W.; Guyatt, G. Deep venous thrombosis in medical-surgical critically ill patients: Prevalence, incidence, and risk factors. Crit. Care Med. 2005, 33, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Williams, M.D. Venous thromboembolism in critically ill patients. Observations from a randomized trial in sepsis. Thromb. Haemost. 2009, 101, 139–144. [Google Scholar] [PubMed]
- Van der Poll, T.; Opal, S.M. Should all septic patients be given systemic anticoagulation? Intensive Care Med. 2017, 43, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Perepu, U.S.; Chambers, I.; Wahab, A.; Ten Eyck, P.; Wu, C.; Dayal, S.; Sutamtewagul, G.; Bailey, S.R.; Rosenstein, L.J.; Lentz, S.R. Standard prophylactic versus intermediate dose enoxaparin in adults with severe COVID-19: A multi-center, open-label, randomized controlled trial. J. Thromb. Haemost. JTH 2021, 19, 2225–2234. [Google Scholar] [CrossRef]
- Sadeghipour, P.; Talasaz, A.H.; Rashidi, F.; Sharif-Kashani, B.; Beigmohammadi, M.T.; Farrokhpour, M.; Sezavar, S.H.; Payandemehr, P.; Dabbagh, A.; et al.; INSPIRATION Investigators Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality Among Patients With COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. JAMA 2021, 325, 1620–1630. [Google Scholar]
- REMAP-CAP Investigators; ACTIV-4a Investigators; ATTACC Investigators. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar] [CrossRef]
- ATTACC Investigators; ACTIV-4a Investigators; REMAP-CAP Investigators. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. JTH 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Brandao, G.M.; Junqueira, D.R.; Rollo, H.A.; Sobreira, M.L. Pentasaccharides for the treatment of deep vein thrombosis. Cochrane Database Syst. Rev. 2017, 12, CD011782. [Google Scholar] [CrossRef]
- Keshari, R.S.; Silasi, R.; Popescu, N.I.; Georgescu, C.; Chaaban, H.; Lupu, C.; McCarty, O.J.T.; Esmon, C.T.; Lupu, F. Fondaparinux pentasaccharide reduces sepsis coagulopathy and promotes survival in the baboon model of Escherichia coli sepsis. J. Thromb. Haemost. JTH 2020, 18, 180–190. [Google Scholar] [CrossRef]
- Savi, P.; Chong, B.H.; Greinacher, A.; Gruel, Y.; Kelton, J.G.; Warkentin, T.E.; Eichler, P.; Meuleman, D.; Petitou, M.; Herault, J.P.; et al. Effect of fondaparinux on platelet activation in the presence of heparin-dependent antibodies: A blinded comparative multicenter study with unfractionated heparin. Blood 2005, 105, 139–144. [Google Scholar] [CrossRef]
- Cardillo, G.; Viggiano, G.V.; Russo, V.; Mangiacapra, S.; Cavalli, A.; Castaldo, G.; Agrusta, F.; Bellizzi, A.; Amitrano, M.; Iannuzzo, M.; et al. Antithrombotic and Anti-Inflammatory Effects of Fondaparinux and Enoxaparin in Hospitalized COVID-19 Patients: The FONDENOXAVID Study. J. Blood Med. 2021, 12, 69–75. [Google Scholar] [CrossRef]
- Beinrohr, L.; Murray-Rust, T.A.; Dyksterhuis, L.; Závodszky, P.; Gál, P.; Pike, R.N.; Wijeyewickrema, L.C. Serpins and the complement system. Methods Enzymol. 2011, 499, 55–75. [Google Scholar]
- Vicci, H.; Eblen-Zajjur, A.; López, M.; Crespo, G.; Navarro, M. Enoxaparin pretreatment effect on local and systemic inflammation biomarkers in the animal burn model. Inflammopharmacology 2019, 27, 521–529. [Google Scholar] [CrossRef]
- Myers, D.D., Jr.; Rectenwald, J.E.; Bedard, P.W.; Kaila, N.; Shaw, G.D.; Schaub, R.G.; Farris, D.M.; Hawley, A.E.; Wrobleski, S.K.; Henke, P.K.; et al. Decreased venous thrombosis with an oral inhibitor of P selectin. J. Vasc. Surg. 2005, 42, 329–336. [Google Scholar] [CrossRef]
- Diaz, J.A.; Wrobleski, S.K.; Alvarado, C.M.; Hawley, A.E.; Doornbos, N.K.; Lester, P.A.; Lowe, S.E.; Gabriel, J.E.; Roelofs, K.J.; Henke, P.K.; et al. P-selectin inhibition therapeutically promotes thrombus resolution and prevents vein wall fibrosis better than enoxaparin and an inhibitor to von Willebrand factor. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 829–837. [Google Scholar] [CrossRef]
- Becattini, C.; Agnelli, G.; Schenone, A.; Eichinger, S.; Bucherini, E.; Silingardi, M.; Bianchi, M.; Moia, M.; Ageno, W.; Vandelli, M.R.; et al. Aspirin for preventing the recurrence of venous thromboembolism. N. Engl. J. Med. 2012, 366, 1959–1967. [Google Scholar] [CrossRef]
- Hess, C.N.; Capell, W.H.; Bristow, M.R.; Ruf, W.; Szarek, M.; Morrow, D.A.; Nicolau, J.C.; Graybill, C.A.; Marshall, D.; Hsia, J.; et al. Rationale and design of a study to assess the safety and efficacy of rNAPc2 in COVID-19: The Phase 2b ASPEN-COVID-19 trial. Am. Heart J. 2022, 246, 136–143. [Google Scholar] [CrossRef]
- Strich, J.R.; Ramos-Benitez, M.J.; Randazzo, D.; Stein, S.R.; Babyak, A.; Davey, R.T.; Suffredini, A.F.; Childs, R.W.; Chertow, D.S. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J. Infect. Dis. 2021, 223, 981–984. [Google Scholar] [CrossRef]
- Kanthi, Y.; Knight, J.S.; Zuo, Y.; Pinsky, D.J. New (re)purpose for an old drug: Purinergic modulation may extinguish the COVID-19 thromboinflammatory firestorm. JCI Insight 2020, 5, 140971. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.; van Rosmalen, J.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef]
- Nickel, K.F.; Long, A.T.; Fuchs, T.A.; Butler, L.M.; Renné, T. Factor XII as a Therapeutic Target in Thromboembolic and Inflammatory Diseases. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 13–20. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mechanism | Pathway |
|---|---|
| Endothelial apoptosis | LPS induces cell death. NETs produce cell death through the direct toxicity of histones and proteases. |
| Endothelial denudation | LPS provokes endothelial cell detachment from basal membrane/internal elastic lamina. |
| Endothelial sensitization | ANGPT-2 sensitizes endothelial cells to inflammatory cytokines and promotes leakage. |
| Endothelial permeability | Inflammatory cytokines increase the EC permeability. |
| Alteration of endothelial histology | LPS can induce nuclear vacuolization, cytoplasmic swelling and protrusion and cytoplasmic fragmentation. |
| Suppression of endothelial anticoagulant receptors | Inflammatory cytokines downregulate EPCR and thrombomodulin. |
| Catecholamines-induced injury | Elevated levels of noradrenaline cause glycocalyx disruption. |
| Reduced sensitivity to catecholamines | LPS reduces vessel relaxation mediated by ACh. |
| Joint proteins internalization | Inflammation induces VE-cadherin dislocation. |
| Function Evaluated | Laboratory Tests |
|---|---|
| Platelets | Platelets count |
| Coagulation | Partial thromboplastin time (PT) and activated partial thromboplastin time (aPTT) |
| Fibrinogen | |
| Anticoagulant markers | Protein C and antithrombin III (AT III) |
| Fibrinolysis markers | D-dimer |
| Fibrinolytic activity | Plasminogen and α2-antiplasmin |
| Antifibrinolytic activity | Plasminogen activator inhibitor 1 (PAI-1) |
| DIC markers | Prothrombin activation fragment Fl and F2, factor IX (FIX), and factor X (FX) activation peptides |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, E.; Maggio, E.; Pomero, F. Venous Thromboembolism in Sepsis: From Bench to Bedside. Biomedicines 2022, 10, 1651. https://doi.org/10.3390/biomedicines10071651
Galli E, Maggio E, Pomero F. Venous Thromboembolism in Sepsis: From Bench to Bedside. Biomedicines. 2022; 10(7):1651. https://doi.org/10.3390/biomedicines10071651
Chicago/Turabian StyleGalli, Eleonora, Elena Maggio, and Fulvio Pomero. 2022. "Venous Thromboembolism in Sepsis: From Bench to Bedside" Biomedicines 10, no. 7: 1651. https://doi.org/10.3390/biomedicines10071651
APA StyleGalli, E., Maggio, E., & Pomero, F. (2022). Venous Thromboembolism in Sepsis: From Bench to Bedside. Biomedicines, 10(7), 1651. https://doi.org/10.3390/biomedicines10071651