
The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy



Abstract
1. Introduction
2. Autophagy and Cancer
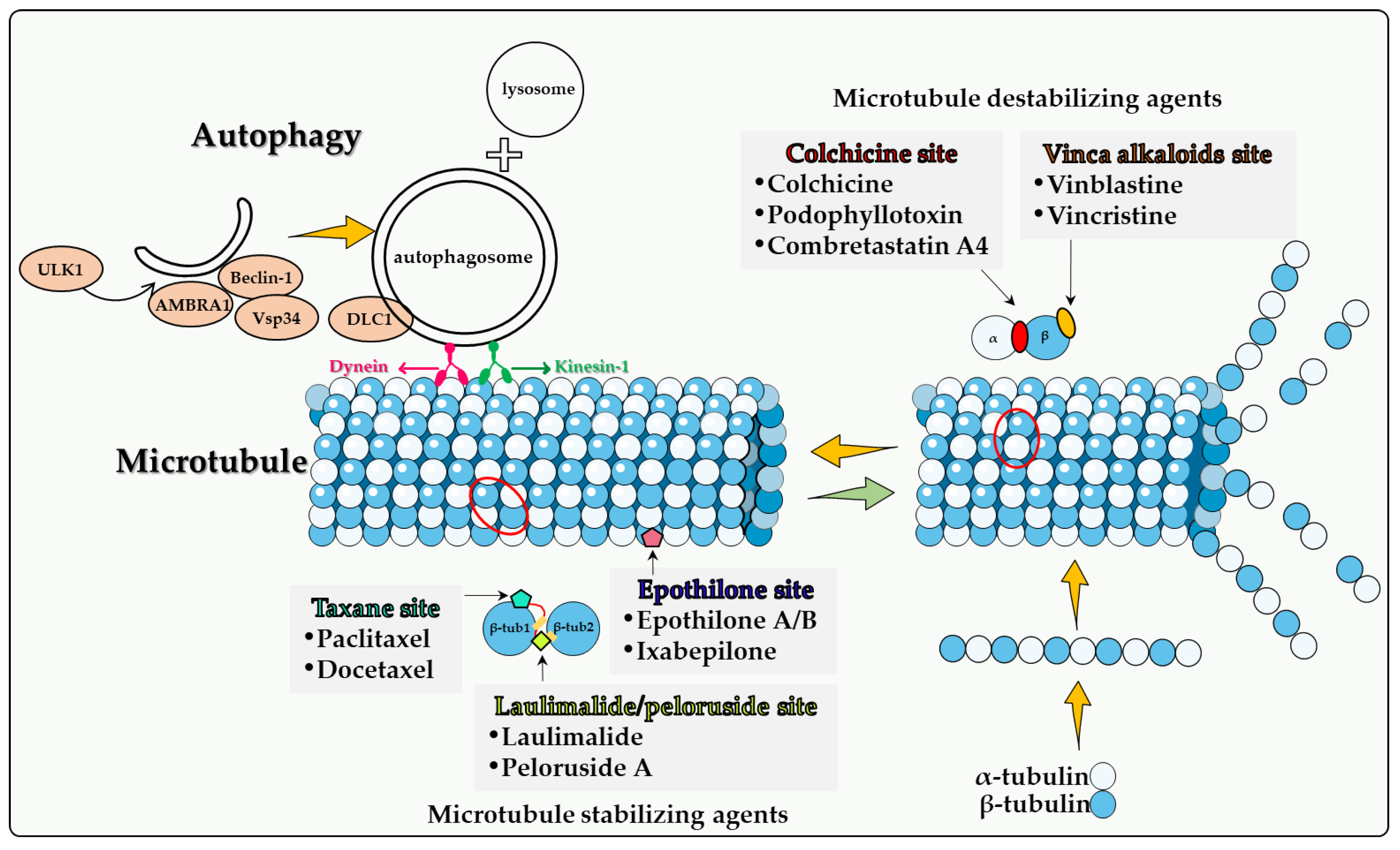
3. Microtubules and Microtubule Poisons
4. Direct Involvement of Microtubules in Autophagy
4.1. Colchicine Site
4.2. Vinca Alkaloid Site
4.3. Taxane Site
4.4. Epothilone Site
4.5. Laulimalide/Peloruside Site
5. Summary and Overview
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gewirtz, D.A. Is autophagy always a barrier to cisplatin therapy? Biomolecules 2022, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Therapeutic Targeting of Autophagy. eBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.; Codogno, P.; Morel, E. ER –plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI 3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef]
- Lystad, A.H.; Carlsson, S.R.; Simonsen, A. Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in autophagy and related processes. Autophagy 2019, 15, 1485–1486. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Ulasov, I.; Fares, J.; Timashev, P.; Lesniak, M.S. Editing cytoprotective autophagy in glioma: An unfulfilled potential for therapy. Trends Mol. Med. 2019, 26, 252–262. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The Autophagy-Senescence Connection in Chemotherapy: Must Tumor Cells (Self) Eat Before They Sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef]
- Patel, N.H.; Xu, J.; Saleh, T.; Wu, Y.; Lima, S.; Gewirtz, D.A. Influence of nonprotective autophagy and the autophagic switch on sensitivity to cisplatin in non-small cell lung cancer cells. Biochem. Pharmacol. 2020, 175, 113896. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Emery, S.M.; Maycotte, P.; Thorburn, A.; Chakradeo, S.; Gewirtz, D.A. Autophagy Inhibition for Chemosensitization and Radiosensitization in Cancer: Do the Preclinical Data Support This Therapeutic Strategy? J. Pharmacol. Exp. Ther. 2013, 344, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Patel, N.H.; Cudjoe, J.E.K.; Alotaibi, M.; Gewirtz, D.A. Studies of non-protective autophagy provide evidence that recovery from therapy-induced senescence is independent of early autophagy. Int. J. Mol. Sci. 2020, 21, 1427. [Google Scholar] [CrossRef] [PubMed]
- Caplow, M. Microtubule dynamics. Curr. Opin. Cell Biol. 1992, 4, 58–65. [Google Scholar] [CrossRef]
- Carlier, M.F.; Hill, T.L.; Chen, Y. Interference of GTP hydrolysis in the mechanism of microtubule assembly: An experimental study. Proc. Natl. Acad. Sci. USA 1984, 81, 771–775. [Google Scholar] [CrossRef]
- Jánosi, I.M.; Chrétien, D.; Flyvbjerg, H. Structural microtubule cap: Stability, catastrophe, rescue, and third state. Biophys. J. 2002, 83, 1317–1330. [Google Scholar] [CrossRef]
- Mitchison, T.J.; Kirschner, M.W. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin superfamily. J. Cell. Sci. 2001, 114, 2723–2733. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Cutts, J.H.; Beer, C.T.; Noble, R.L. Biological properties of Vincaleukoblastine, an alkaloid in Vinca rosea Linn, with reference to its antitumor action. Cancer Res. 1960, 20, 1023–1031. [Google Scholar]
- Schneider, F.; Pan, L.; Ottenbruch, M.; List, T.; Gaich, T. The chemistry of nonclassical taxane diterpene. Acc. Chem. Res. 2021, 54, 2347–2360. [Google Scholar] [CrossRef]
- Yang, C.H.; Horwitz, S.B. Taxol(R): The first microtubule stabilizing agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef]
- Huitorel, P. From cilia and flagella to intracellular motility and back again: A review of a few aspects of microtubule-based motility. Biol. Cell 1988, 63, 249–258. [Google Scholar] [CrossRef]
- Forth, S.; Kapoor, T.M. The mechanics of microtubule networks in cell division. J. Cell Biol. 2017, 216, 1525–1531. [Google Scholar] [CrossRef]
- Tangutur, A.D.; Kumar, D.; Krishna, K.V.; Kantevari, S. Microtubule targeting agents as cancer chemotherapeutics: An overview of molecular hybrids as stabilizing and destabilizing agents. Curr. Top. Med. Chem. 2017, 17, 2523–2537. [Google Scholar] [CrossRef]
- Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 2–106. [Google Scholar] [CrossRef]
- Hari, M.; Yang, H.; Zeng, C.; Canizales, M.; Cabral, F. Expression of class III beta-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil. Cytoskelet. 2003, 56, 45–56. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent Movement of Autophagosomes Mediates Efficient Encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Poüs, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194. [Google Scholar] [CrossRef]
- Luo, S.; Garcia-Arencibia, M.; Zhao, R.; Puri, C.; Toh, P.P.; Sadiq, O.; Rubinsztein, D.C. Bim Inhibits autophagy by recruiting beclin 1 to microtubules. Mol. Cell 2012, 47, 359–370. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunra, L. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The cytoskeleton–autophagy connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006, 281, 36303–36316. [Google Scholar] [CrossRef]
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Nowosad, A.; Creff, J.; Jeannot, P.; Culerrier, R.; Codogno, P.; Manenti, S.; Nguyen, L.; Besson, A. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis. 2021, 12, 481. [Google Scholar] [CrossRef]
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Das, A.; Datta, S.; Ganguli, A.; Chakrabarti, G. Colchicine induces autophagy and senescence in lung cancer cells at clinically admissible concentration: Potential use of colchicine in combination with autophagy inhibitor in cancer therapy. Tumor Biol. 2016, 37, 10653–10664. [Google Scholar] [CrossRef]
- Sivakumar, G. Colchicine semisynthetics: Chemotherapeutics for cancer? Curr. Med. Chem. 2013, 20, 892–898. [Google Scholar] [PubMed]
- Arthur, C.R.; Gupton, J.T.; Kellogg, G.; Yeudall, W.A.; Cabot, M.C.; Newsham, I.F.; Gewirtz, D.A. Autophagic cell death, polyploidy and senescence induced in breast tumor cells by the substituted pyrrole JG-03-14, a novel microtubule poison. Biochem. Pharmacol. 2007, 74, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Biggers, J.W.; Nguyen, T.; Di, X.; Gupton, J.T.; Henderson, S.C.; Emery, S.M.; Alotaibi, M.; White, K.L., Jr.; Brown, R.; Almenara, J.; et al. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03-14. Cancer Chemother. Pharmacol. 2013, 71, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Larocque, K.; Ovadje, P.; Djurdjevic, S.; Mehdi, M.; Green, J.; Pandey, S. Novel analogue of colchicine induces selective pro-death autophagy and necrosis in human cancer cells. PLoS ONE 2014, 9, e87064. [Google Scholar] [CrossRef]
- Fang, K.-M.; Liu, J.-J.; Li, C.-C.; Cheng, C.-C.; Hsieh, Y.-T.; Chai, K.M.; Lien, Y.-A.; Tzeng, S.-F. Colchicine derivative as a potential anti-glioma compound. J. Neuro-Oncol. 2015, 124, 403–412. [Google Scholar] [CrossRef]
- Fan, H.-Y.; Zhu, Z.-L.; Xian, H.-C.; Wang, H.-F.; Chen, B.-J.; Tang, Y.-J.; Liang, X.-H. Insight into the molecular mechanism of podophyllotoxin derivatives as anticancer drugs. Front. Cell Dev. Biol. 2021, 9, 709075. [Google Scholar] [CrossRef]
- Zhao, W.; Cong, Y.; Li, H.M.; Li, S.; Shen, Y.; Qi, Q.; Zhang, Y.; Li, Y.-Z.; Tang, Y.-J. Challenges and potential for improving the druggability of podophyllotoxin-derived drugs in cancer chemotherapy. Nat. Prod. Rep. 2021, 38, 470–488. [Google Scholar] [CrossRef]
- Cortese, F.; Bhattacharyya, B.; Wolff, J. Podophyllotoxin as a probe for the colchicine binding site of tubulin. J. Biol. Chem. 1977, 252, 1134–1140. [Google Scholar] [CrossRef]
- Stähelin, H.F.; Von Wartburg, A. The chemical and biological route from podophyllotoxin glucoside to etoposide: Ninth Cain memorial Award lecture. Cancer Res. 1991, 51, 5–15. [Google Scholar]
- Choi, J.Y.; Hong, W.G.; Cho, J.H.; Kim, E.M.; Kim, J.; Jung, C.-H.; Hwang, S.-G.; Um, H.-D.; Park, J.K. Podophyllotoxin acetate triggers anticancer effects against non-small cell lung cancer cells by promoting cell death via cell cycle arrest, ER stress and autophagy. Int. J. Oncol. 2015, 47, 1257–1265. [Google Scholar] [CrossRef]
- Wang, J.; Long, L.; Chen, Y.; Xu, Y.; Zhang, L. Design, synthesis and antineoplastic activity of novel hybrids of podophyllotoxin and indirubin against human leukaemia cancer cells as multifunctional anti-MDR agents. Bioorg. Med. Chem. Lett. 2018, 28, 1817–1824. [Google Scholar] [CrossRef]
- Ren, J.; Liu, Y.; Li, L.; Zhao, Y.; Li, Z.; Wu, C.; Chen, L.; Hu, K. OAMDP, a novel podophyllotoxin derivative, induces apoptosis, cell cycle arrest and autophagy in hepatoma HepG2 cells. Cell Biol. Int. 2018, 42, 194–204. [Google Scholar] [CrossRef]
- Karatoprak, G.Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakcı, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An overview of structure, probable mechanisms of action and potential applications. Molecules 2020, 25, 2560. [Google Scholar] [CrossRef]
- Dowlati, A.; Robertson, K.; Cooney, M.; Petros, W.P.; Stratford, M.; Jesberger, J.; Rafie, N.; Overmoyer, B.; Makkar, V.; Stambler, B.; et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002, 62, 3408–3416. [Google Scholar]
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T.; et al. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152. [Google Scholar] [CrossRef]
- West, C.M.; Price, P. Combretastatin A4 phosphate. Anticancer. Drugs 2004, 15, 179–187. [Google Scholar] [CrossRef]
- Li, Y.; Luo, P.; Wang, J.; Dai, J.; Yang, X.; Wu, H.; Yang, B.; He, Q. Autophagy blockade sensitizes the anticancer activity of CA-4 via JNK-Bcl-2 pathway. Toxicol. Appl. Pharmacol. 2014, 274, 319–327. [Google Scholar] [CrossRef]
- Greene, L.M.; O’Boyle, N.M.; Nolan, D.P.; Meegan, M.J.; Zisterer, D.M. The vascular targeting agent Combretastatin-A4 directly induces autophagy in adenocarcinoma-derived colon cancer cells. Biochem. Pharmacol. 2012, 84, 612–624. [Google Scholar] [CrossRef]
- Wang, H.; Li, W.; Xu, J.; Zhang, T.; Zuo, D.; Zhou, Z.; Lin, B.; Wang, G.; Wang, Z.; Sung, W.; et al. NDRG1 inhibition sensitizes osteosarcoma cells to combretastatin A-4 through targeting autophagy. Cell Death Dis. 2017, 8, e3048. [Google Scholar] [CrossRef]
- Rustin, G.J.; Galbraith, S.M.; Anderson, H.; Stratford, M.; Folkes, L.K.; Sena, L.; Gumbrell, L.; Price, P.M. Phase I clinical trial of weekly combretastatin A4 phosphate: Clinical and pharmacokinetic results. J. Clin. Oncol. 2003, 21, 2815–2822. [Google Scholar] [CrossRef]
- Hoang, V.C.; Chow, A.; Emmenegger, U. Abstract 1688: Autophagy inhibition enhances the antitumor effects of combretastatin A4 phosphate (CA4P). Cancer Res. 2013, 73 (Suppl. S8), 1688. [Google Scholar] [CrossRef]
- Greene, L.M.; Meegan, M.J.; Zisterer, D. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Agrawal, K. Vinblastine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar]
- Lee, C.-T.; Huang, Y.-W.; Yang, C.-H.; Huang, K.-S. Drug delivery systems and combination therapy by using vinca alkaloids. Curr. Top. Med. Chem. 2015, 15, 1491–1500. [Google Scholar] [CrossRef]
- Downing, K.H. Structural Basis for the Interaction of Tubulin with Proteins and Drugs that Affect Microtubule Dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef]
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar]
- Silvestri, R. New prospects for vinblastine analogues as anticancer agents. J. Med. Chem. 2013, 56, 625–627. [Google Scholar] [CrossRef]
- Carlson, R.O. New tubulin targeting agents currently in clinical development. Expert Opin. Investig. Drugs 2008, 17, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Punnonen, E.-L.; Reunanen, H. Effects of vinblastine, leucine, and histidine, and 3-methyladenine on autophagy in ehrlich ascites cells. Exp. Mol. Pathol. 1990, 52, 87–97. [Google Scholar] [CrossRef]
- Xie, R.; Nguyen, S.; McKeehan, W.L.; Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010, 11, 89. [Google Scholar] [CrossRef]
- Adiseshaiah, P.P.; Clogston, J.D.; McLeland, C.B.; Rodriguez, J.; Potter, T.M.; Neun, B.W.; Skoczen, S.L.; Shanmugavelandy, S.S.; Kester, M.; Stern, S.T.; et al. Synergistic combination therapy with nanoliposomal C6-ceramide and vinblastine is associated with autophagy dysfunction in hepatocarcinoma and colorectal cancer models. Cancer Lett. 2013, 337, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar] [CrossRef] [PubMed]
- Belounis, A.; Nyalendo, C.; Le Gall, R.; Imbriglio, T.V.; Mahma, M.; Teira, P.; Beaunoyer, M.; Cournoyer, S.; Haddad, E.; Vassal, G.; et al. Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 2016, 16, 891. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Hui, W.; Li, H.; Wang, Z.; Guo, C.; Peng, R.; Gu, J.; Chen, Y.; Ouyang, Q. Discovery of novel autophagy inhibitors and their sensitization abilities for vincristine-resistant esophageal cancer cell line Eca109/VCR. Chem. Med. Chem. 2020, 15, 970–981. [Google Scholar] [CrossRef]
- Sheikh‐Zeineddini, N.; Safaroghli-Azar, A.; Salari, S.; Bashash, D. C-Myc inhibition sensitizes pre-B ALL cells to the anti-tumor effect of vincristine by altering apoptosis and autophagy: Proposing a probable mechanism of action for 10058-F4. Eur. J. Pharmacol. 2020, 870, 172821. [Google Scholar] [CrossRef]
- Sun, J.; Feng, D.; Xi, H.; Luo, J.; Zhou, Z.; Liu, Q.; Chen, Y.; Shao, Q. CD24 blunts the sensitivity of retinoblastoma to vincristine by modulating autophagy. Mol. Oncol. 2020, 14, 1740–1759. [Google Scholar] [CrossRef]
- Li, Z.; Wang, N.; Yue, T.; Liu, L. Matrine reverses the drug resistance of K562/ADM cells to ADM and VCR via promoting autophagy. Transl. Cancer Res. 2020, 9, 786–794. [Google Scholar] [CrossRef]
- Takahashi, T.; Honma, Y.; Miyake, T.; Adachi, K.; Takami, S.; Okada, M.; Kumanomidou, S.; Ikejiri, F.; Jo, Y.; Onishi, C.; et al. Synergistic combination therapy with cotylenin A and vincristine in multiple myeloma models. Int. J. Oncol. 2015, 46, 1801–1809. [Google Scholar] [CrossRef]
- Schiff, P.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136. [Google Scholar] [CrossRef]
- A Jordan, M.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. 1993, 90, 9552–9556. [Google Scholar] [CrossRef]
- Raveendran, R.S.; Baby, S. Resistance to intervention: Paclitaxel in breast cancer. Mini-Rev. Med. Chem. 2021, 21, 1237–1268. [Google Scholar]
- Baird, R.D.; Tan, D.S.P.; Kaye, S.B. Weekly paclitaxel in the treatment of recurrent ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 575–582. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, S.; Liu, S.; Wang, Y.; Liu, G. Emerging nanomedicines of paclitaxel for cancer treatment. J. Control. Release 2022, 342, 280–294. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. J. Am. Med. Assoc. 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Zou, C.-F.; Jia, L.; Jin, H.; Yao, M.; Zhao, N.; Huan, J.; Lu, Z.; Bast, R.C.; Feng, Y.; Yu, Y. Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer 2011, 11, 22. [Google Scholar] [CrossRef]
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746. [Google Scholar] [CrossRef]
- Zhang, S.-F.; Wang, X.-Y.; Fu, Z.-Q.; Peng, Q.-H.; Zhang, J.-Y.; Ye, F.; Fu, Y.-F.; Zhou, C.-Y.; Lu, W.-G.; Cheng, X.-D.; et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 2015, 11, 225–238. [Google Scholar] [CrossRef]
- Xi, G.; Hu, X.; Wu, B.; Jiang, H.; Young, C.Y.; Pang, Y.; Yuan, H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011, 307, 141–148. [Google Scholar] [CrossRef]
- Karasic, T.B.; O′Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, A.D.; et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Anand, K.; Niravath, P.; Patel, T.; Ensor, J.; Rodriguez, A.; Boone, T.; Wong, S.T.; Chang, J.C. A phase II study of the efficacy and safety of chloroquine in combination with taxanes in the treatment of patients with advanced or metastatic anthracycline-refractory breast cancer. Clin. Breast Cancer 2021, 21, 199–204. [Google Scholar] [CrossRef]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef]
- Cristofani, R.; Marelli, M.M.; Cicardi, M.E.; Fontana, F.; Marzagalli, M.; Limonta, P.; Poletti, A.; Moretti, R.M. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018, 9, 889. [Google Scholar] [CrossRef]
- Pickard, R.D.; Spencer, B.H.; McFarland, A.J.; Bernaitis, N.; Davey, A.K.; Perkins, A.V.; Chess-Williams, R.; McDermott, C.M.; Forbes, A.; Christie, D.; et al. Paradoxical effects of the autophagy inhibitor 3-methyladenine on docetaxel-induced toxicity in PC-3 and LNCaP prostate cancer cells. Naunyn-Schmiedebergs Arch. Fur Exp. Pathol. Und Pharmakol. 2015, 388, 793–799. [Google Scholar] [CrossRef]
- Gerth, K.; Bedorf, N.; Höfle, G.; Irschik, H.; Reichenbach, H. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. 1996, 49, 560–563. [Google Scholar] [CrossRef]
- Nettles, J.H.; Downing, K.H. The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science 2004, 305, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Zhang, X.-G.; Harris, C.R.; Kuduk, S.D.; Balog, A.; Savin, K.A.; Bertino, J.R.; Danishefsky, S.J. Desoxyepothilone B is curative against human tumor xenografts that are refractory to paclitaxel. Proc. Natl. Acad. Sci. USA 1998, 95, 15798–15802. [Google Scholar] [CrossRef] [PubMed]
- Fumoleau, P.; Coudert, B.; Isambert, N.; Ferrant, E. Novel tubulin-targeting agents: Anticancer activity and pharmacologic profile of epothilones and related analogues. Ann. Oncol. 2007, 18 (Suppl. S5), v9–v15. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kepp, O.; Martins, I.; Vitale, I.; Souquère, S.; Castedo, M.; Pierron, G.; Kroemer, G. Defective autophagy associated with LC3 puncta in epothilone-resistant cancer cells. Cell Cycle 2010, 9, 377–383. [Google Scholar] [CrossRef]
- Oehler, C.; Von Bueren, A.O.; Furmanova, P.; Broggini-Tenzer, A.; Orlowski, K.; Rutkowski, S.; Frei, K.; Grotzer, M.A.; Pruschy, M. The microtubule stabilizer patupilone (epothilone B) is a potent radiosensitizer in medulloblastoma cells. Neuro Oncol. 2011, 13, 1000–1010. [Google Scholar] [CrossRef][Green Version]
- Rogalska, A.; Gajek, A.; Marczak, A. Suppression of autophagy enhances preferential toxicity of epothilone A and epothilone B in ovarian cancer cells. Phytomedicine 2019, 61, 152847. [Google Scholar] [CrossRef]
- Rugo, H.S.; Barry, W.T.; Moreno-Aspitia, A.; Lyss, A.P.; Cirrincione, C.; Leung, E.; Mayer, E.L.; Naughton, M.; Toppmeyer, D.; Carey, L.A.; et al. Randomized phase III trial of paclitaxel once per week compared with nanoparticle albumin-bound nab-paclitaxel once per week or ixabepilone with bevacizumab as first-line chemotherapy for locally recurrent or metastatic breast cancer: CALGB 40502/NCCTG N063H (Alliance). J. Clin. Oncol. 2015, 33, 2361–2369. [Google Scholar]
- Tanei, T.; Choi, D.S.; Rodriguez, A.A.; Liang, D.H.; E Dobrolecki, L.; Ghosh, M.; Landis, M.D.; Chang, J.C. Antitumor activity of Cetuximab in combination with Ixabepilone on triple negative breast cancer stem cells. Breast Cancer Res. 2016, 18, 6. [Google Scholar] [CrossRef]
- Cao, D.-S.; Jiang, S.-L.; Guan, Y.-D.; Chen, X.-S.; Zhang, L.-X.; Zhang, Y.; Chen, A.F.; Yang, J.-M.; Cheng, Y. A multi-scale systems pharmacology approach uncovers the anti-cancer molecular mechanism of Ixabepilone. Eur. J. Med. Chem. 2020, 199, 112421. [Google Scholar] [CrossRef]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- West, L.M.; Northcote, P.T.; Battershill, C.N. Peloruside A: A Potent Cytotoxic Macrolide Isolated from the New Zealand Marine Sponge Mycale sp. J. Org. Chem. 2000, 65, 445–449. [Google Scholar] [CrossRef]
- Gaitanos, T.N.; Buey, R.M.; Díaz, J.F.; Northcote, P.T.; Teesdale-Spittle, P.; Andreu, J.M.; Miller, J.H. Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004, 64, 5063–5067. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Pineda, O.; Vilarrasa, J. Further insight into the interactions of the cytotoxic macrolides laulimalide and peloruside a with their common binding site. ACS Omega 2018, 3, 1770–1782. [Google Scholar] [CrossRef]
- Gollner, A.; Altmann, K.-H.; Gertsch, J.; Mulzer, J. The laulimalide family: Total synthesis and biological evaluation of neolaulimalide, isolaulimalide, laulimalide and a nonnatural analogue. Chem. A Eur. J. 2009, 15, 5979–5997. [Google Scholar] [CrossRef]


{kind=link}
| Agents | Cancer Type | Autophagy | Autophagy Inhibitor | Autophagy and Cell Death | Ref. |
|---|---|---|---|---|---|
| Colchicine | A549 lung cancer cell line | Autophagy induction (associated with senescence) | 3-MA | Cytoprotective autophagy | [50] |
| Colchicine derivative “JG-03-14” | MCF-7 and MDA-MB-231 breast cancer cells | Autophagy induction | N/A | Cytotoxic autophagy | [52] |
| Colchicine derivative “JG-03-14” | B16/F10 melanoma and HCT-116 colon cancer cells | Autophagy induction | CQ and Baf A1 | Cytotoxic autophagy | [53] |
| Colchicine derivative “Green 1” | PANC-1 pancreatic cancer and E6-1 or Jurkat acute T cell leukemia cell lines | Autophagy induction | N/A | Cytotoxic autophagy | [54] |
| Colchicine derivative “AD1” | U87MG and U373MG human malignant glioblastoma cell lines | Autophagy induction | N/A | Cytotoxic autophagy | [55] |
| Podophyllotoxin acetate | A549 and NCI-H1299 human non-small cell lung cancer cell lines | Autophagy induction | N/A | Cytotoxic autophagy | [60] |
| Podophyllotoxin derivative “Da-1” | K562/VCR chronic myeloid leukemia cell lines | Autophagy induction | N/A | Cytotoxic autophagy | [61] |
| Podophyllotoxin derivative “OAMDP” | HepG2 hepatoma cell line | Autophagy induction | N/A | Cytotoxic autophagy | [62] |
| Combretastatin A-4 | MDA-MB-231 breast tumor cells, SGC-7901 human gastric tumor cells and SMMC-7721 human hepatocellular carcinoma cells | Autophagy induction | In vitro; 3-MA/Baf A1/siRNAs against Atg5 and Beclin 1 genes/BCL2 inhibitor (ABT-737) JNK inhibitor or JNK siRNA In vivo; 3-MA | Cytoprotective autophagy | [67] |
| Combretastatin A-4 | CT-26 and HT-29 adenocarcinoma cell lines | Autophagy induction | 3-MA, Baf A1 | Non-protective autophagy in CT-26 cell line, Cytoprotective autophagy in HT-29 cell line | [68] |
| Combretastatin A-4 | SJSA and MG63 human osteosarcoma cell lines | Autophagy induction | CQ | Cytoprotective autophagy | [69] |
| Combretastatin A-4 phosphate | PC3 prostate cancer xenografts | Autophagy induction | autophagy-defective PC3 prostate cancer xenografts (developed with retrovirally transducing PC-3 cells with ATG4BC74A) | Cytoprotective autophagy | [71,72] |
| Vinblastine | Ehrlich ascites tumor cells | Autophagy inhibition with autophagic vacuoles accumulation | N/A | N/A | [80] |
| Vinblastine | HepG2 human hepatocarcinoma and LS174T colon cancer cell lines | blocked autophagy maturation | CQ | Autophagy inhibition | [82] |
| Vincristine | Neuroblastoma cell lines | Autophagy induction | In vivo; ATG5 knockdown In vivo; HCQ | Cytoprotective autophagy | [84] |
| Vincristine | Eca-109/VCR esophagus cancer cell line | Autophagy induction | autophagy inhibitor “4d” | Cytoprotective autophagy | [85] |
| Vincristine | REH and Nalm-6 pre-B acute lymphoblastic leukemia (ALL) cell lines | Autophagy induction | CQ | Cytoprotective autophagy | [86] |
| Vincristine | human retinoblastoma cells | Autophagy induction | CQ, CD24 knockdown | Cytoprotective autophagy | [87] |
| Vincristine | K562/ADM cell line | Autophagy induction | CQ reversed the inhibitory effect of MAT | MAT promote cytotoxic autophagy | [88] |
| Vincristine | multiple myeloma cells | Autophagy induction | N/A | Cotylenin A promote cytotoxic autophagy | [89] |
| Paclitaxel | breast cancer cells | Paclitaxel prevents autophagosome maturation and lysosome fusion in breast cancer cells | 3-MA | Activates apoptosis as a result of autophagic flux inhibition in cancer cells | [98] |
| Paclitaxel | breast cancer cells | Paclitaxel alone did not induce autophagy in breast cancer cells, it enhanced ARHI-induced autophagy. | N/A | When ARHI was re-expressed in breast cancer cells treated with paclitaxel, the growth inhibitory effect of paclitaxel was enhanced in both the cell culture and the xenografts. | [97] |
| Paclitaxel | A2780, 3AO, and SKOV3 ovarian cancer cells | Paclitaxel increases autophagosome formation and autophagic flux | Beclin-1 deficiency | Cytoprotective autophagy | [99] |
| Paclitaxel | A549 cells; U87, PC3 and HT-29 cells | Autophagy induction (no p62/SQSTM1 detected) | 3-MA /Beclin 1 siRNA | Cytoprotective autophagy | [100] |
| Docetaxel | LNCaP, PC3, and DU145 | Autophagy induction | 3-MA | Non-protective autophagy | [105] |
| Docetaxel | docetaxel resistant prostate cancer cell lines | Autophagy induction | N/A | Non-protective autophagy | [104] |
| Docetaxel | prostate cancer cells | Autophagy induction | 3-MA | Cytoprotective autophagy | [106] |
| Epothilone A and Epothilone B | ovarian cancer cells | Autophagy induction | Baf A1 | Cytoprotective autophagy | [113] |
| Ixabepilone | hepatic carcinoma, glioma cells and breast cancer cells | Autophagy induction | In vitro: CQ, 3-MA, beclin-1 si RNA In vivo: CQ | Cytoprotective autophagy | [116] |
| Ixabepilone | MDA-MB-231 and SUM159 cells | Ixabepilone increased p62/SQSTM1 expression, Ixabepilone either reduced or had no effect on the basal levels of LC3b-II | N/A | N/A | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Elshazly, A.M.; Gewirtz, D.A. The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy. Biomedicines 2022, 10, 1632. https://doi.org/10.3390/biomedicines10071632
Xu J, Elshazly AM, Gewirtz DA. The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy. Biomedicines. 2022; 10(7):1632. https://doi.org/10.3390/biomedicines10071632
Chicago/Turabian StyleXu, Jingwen, Ahmed M. Elshazly, and David A. Gewirtz. 2022. "The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy" Biomedicines 10, no. 7: 1632. https://doi.org/10.3390/biomedicines10071632
APA StyleXu, J., Elshazly, A. M., & Gewirtz, D. A. (2022). The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy. Biomedicines, 10(7), 1632. https://doi.org/10.3390/biomedicines10071632