
The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis



Abstract
:1. Introduction
2. Osteoarthritis Epidemiology
3. Introduction to Neutrophils and Macrophages Involved in Osteoarthritis
4. Normal Bone and Cartilage Formation and Remodeling
5. Osteoarthritis Pathophysiology
5.1. Cartilage Degradation
5.2. Synovial Tissue
5.3. Chondrogenic Progenitor Cells
5.4. MicroRNAs
5.5. Exosomes
6. Current Treatments & Investigations
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, G.; Jing, W.; Bi, Y.; Li, Y.; Ma, L.; Yang, H.; Zhang, Y. Neutrophil Elastase Induces Chondrocyte Apoptosis and Facilitates the Occurrence of Osteoarthritis via Caspase Signaling Pathway. Front Pharmacol. 2021, 12, 666162. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.D.; Chubinskaya, S.; Guilak, F.; Martin, J.A.; Oegema, T.R.; Olson, S.A.; Buckwalter, J.A. Post-traumatic osteoarthritis: Improved understanding and opportunities for early intervention. J. Orthop. Res. 2011, 29, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines involved in osteoarthritis pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, M.F.; Zhang, X.; Wellman, S.S.; Bolognesi, M.P.; Kraus, V.B. Synergistic roles of macrophages and neutrophils in osteoarthritis progression. Arthritis Rheumatol. 2021, 73, 89–99. [Google Scholar] [CrossRef]
- Fattori, V.; Amaral, F.A.; Verri, W.A. Neutrophils and arthritis: Role in disease and pharmacological perspectives. Pharmacol. Res. 2016, 112, 84–98. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Falconer, A.M.D.; Wright, H.L.; Lin, H.; Yamamoto, K.; Cheung, K.; Charlton, S.H.; Arques, M.D.C.; Janciauskiene, S.; Refaie, R.; et al. Matrix metalloproteinase-13 is fully activated by neutrophil elastase and inactivates its serpin inhibitor, alpha-1 antitrypsin: Implications for osteoarthritis. FEBS J. 2022, 289, 121–139. [Google Scholar] [CrossRef]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef]
- Abramoff, B.; Caldera, F.E. Osteoarthritis. Med. Clin. N. Am. 2020, 104, 293–311. [Google Scholar] [CrossRef]
- Sacitharan, P.K. Ageing and Osteoarthritis. Subcell Biochem. 2019, 91, 123–159. [Google Scholar]
- Xia, B.; Chen, D.; Zhang, J.; Hu, S.; Jin, H.; Tong, P. Osteoarthritis Pathogenesis: A Review of Molecular Mechanisms. Calcif. Tissue Int. 2014, 95, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, T.W.; McCabe, P.S.; McBeth, J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Naimark, A.; Anderson, J.; Kazis, L.; Castelli, W.; Meenan, R.F. The prevalence of knee osteoarthritis in the elderly. the framingham osteoarthritis study. Arthritis Rheum. 1987, 30, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Leyland, K.M.; Hart, D.J.; Javaid, M.K.; Judge, A.; Kiran, A.; Soni, A.; Goulston, L.M.; Cooper, C.; Spector, T.D.; Arden, N.K. The natural history of radiographic knee osteoarthritis: A fourteen-year population-based cohort study. Arthritis Rheum. 2012, 64, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.M.; Helmick, C.G.; Renner, J.B.; Luta, G.; Dragomir, A.D.; Woodard, J.; Fang, F.; Schwartz, T.A.; Nelson, A.E.; Abbate, L.M.; et al. Prevalence of Hip Symptoms and Radiographic and Symptomatic Hip Osteoarthritis in African Americans and Caucasians: The Johnston County Osteoarthritis Project. J. Rheumatol. 2009, 36, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Alhambra, D.; Judge, A.; Javaid, M.K.; Cooper, C.; Diez-Perez, A.; Arden, N.K. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: Influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 2014, 73, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Cameron, K.L.; Driban, J.B.; Svoboda, S.J. Osteoarthritis and the Tactical Athlete: A Systematic Review. J. Athl. Train. 2016, 51, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Driban, J.B.; Hootman, J.M.; Sitler, M.R.; Harris, K.P.; Cattano, N.M. Is Participation in Certain Sports Associated with Knee Osteoarthritis? A Systematic Review. J. Athl. Train. 2017, 52, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Kaneva, M.K. Neutrophil elastase and its inhibitors—Overlooked players in osteoarthritis. FEBS J. 2022, 289, 113–116. [Google Scholar] [CrossRef]
- Kriegova, E.; Manukyan, G.; Mikulkova, Z.; Gabcova, G.; Kudelka, M.; Gajdos, P.; Gallo, J. Gender-related differences observed among immune cells in synovial fluid in knee osteoarthritis. Osteoarthr. Cartil. 2018, 26, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Büyükavcı, R.; Aktürk, S.; Sağ, S. Comparison of blood platelet distribution width and neutrophil-lymphocyte ratio in patients with different grades of knee osteoarthritis. J. Back Musculoskelet. Rehabil. 2018, 31, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Tao, J.; Bae, Y.; Jiang, M.M.; Bertin, T.; Chen, Y.; Yang, T.; Lee, B. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J. Bone Miner. Res. 2013, 28, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Chen, S.; Lee, B. Alteration of Notch signaling in skeletal development and disease. Ann. N. Y. Acad. Sci. 2010, 1192, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteoblast physiology in normal and pathological conditions. Cell Tissue Res. 2011, 343, 289–302. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [Green Version]
- Man, G.S.; Mologhianu, G. Osteoarthritis pathogenesis—A complex process that involves the entire joint. J. Med. Life. 2014, 7, 37–41. [Google Scholar]
- Arden, N.; Blanco, F.; Cooper, C.; Guermazi, A.; Hayashi, D.; Hunter, D.; Javaid, M.K.; Rannou, F.; Roemer, F.; Reginsteret, J.-Y. Atlas of Osteoarthritis; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Kaneva, M.K.; Muley, M.M.; Krustev, E.; Reid, A.R.; Souza, P.R.; Dell’Accio, F.; McDougall, J.J.; Perretti, M. Alpha-1-antitrypsin reduces inflammation and exerts chondroprotection in arthritis. FASEB J. 2021, 35, e21472. [Google Scholar] [CrossRef]
- Weber, A.; Chan, P.M.B.; Wen, C. Do immune cells lead the way in subchondral bone disturbance in osteoarthritis? Prog. Biophys. Mol. Biol. 2019, 148, 21–31. [Google Scholar] [CrossRef]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Haaland, B.; Huebner, J.L.; Wong, S.; Tjai, M.; Wang, C.; Chowbay, B.; Thumboo, J.; Chakraborty, B.; Tan, M.H.; et al. Colchicine lack of effectiveness in symptom and inflammation modification in knee osteoarthritis (COLKOA): A randomized controlled trial. Osteoarthr. Cartil. 2018, 26, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, N.; Dimitriou, I.D.; Rottapel, R. Go with the flow: GEF-H1 mediated shear stress mechanotransduction in neutrophils. Small GTPases 2020, 11, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraden, C.A.; Huebner, J.L.; Hsueh, M.F.; Li, Y.J.; Kraus, V.B. Synovial fluid biomarkers associated with osteoarthritis severity reflect macrophage and neutrophil related inflammation. Arthritis Res. Ther. 2019, 21, 146. [Google Scholar] [CrossRef] [Green Version]
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739. [Google Scholar] [CrossRef] [Green Version]
- De Luca, P.; Kouroupis, D.; Viganò, M.; Perucca-Orfei, C.; Kaplan, L.; Zagra, L.; de Girolamo, L.; Correa, D.; Colombini, A. Human Diseased Articular Cartilage Contains a Mesenchymal Stem Cell-Like Population of Chondroprogenitors with Strong Immunomodulatory Responses. J. Clin. Med. 2019, 8, 423. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef]
- Hamilton, J.L.; Nagao, M.; Levine, B.R.; Chen, D.; Olsen, B.R.; Im, H.-J. Targeting VEGF and Its Receptors for the Treatment of Osteoarthritis and Associated Pain. J. Bone Miner. Res. 2016, 31, 911–924. [Google Scholar] [CrossRef]
- Kasten, K.R.; Prakash, P.S.; Unsinger, J.; Goetzman, H.S.; England, L.G.; Cave, C.M.; Seitz, A.P.; Mazuski, C.N.; Zhou, T.T.; Morre, M.; et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through γδ T-cell IL-17 production in a murine model of sepsis. Infect Immun. 2010, 78, 4714–4722. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Seol, D.; McCabe, D.J.; Choe, H.; Zheng, H.; Yu, Y.; Jang, K.; Walter, M.W.; Lehman, A.D.; Ding, L.; Buckwalter, J.A.; et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012, 64, 3626–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegger, J.; Palm, H.G.; Brenner, R.E. The functional role of chondrogenic stem/progenitor cells: Novel evidence for immunomodulatory properties and regenerative potential after cartilage injury. Eur. Cells Mater. 2018, 36, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Lehmann, C.; Bode, C.; Miosge, N.; Schubert, A. High Mobility Group Box 1 Protein in Osteoarthritic Knee Tissue and Chondrogenic Progenitor Cells: An Ex Vivo and In Vitro Study. Cartilage 2021, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cassatella, M.A. Production of Cytokines By Polymorphonuclear Neutrophils. In Neutrophils New Outlook Old Cells; Imperial College Press: London, UK, 1999; pp. 151–229. [Google Scholar]
- Coutinho de Almeida, R.; Ramos, Y.F.M.; Mahfouz, A.; Den Hollander, W.; Lakenberg, N.; Houtman, E.; Van Hoolwerff, M.; Suchiman, H.E.D.; Ruiz, R.A.; Slagboom, P.E.; et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis. 2019, 78, 270–277. [Google Scholar] [CrossRef]
- Gu, R.; Liu, N.; Luo, S.; Huang, W.; Zha, Z.; Yang, J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine 2016, 95, e4315. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, S.; Jia, Q.; Zhang, A.; Li, Y.; Zhu, Y.; Lv, S.; Zhang, J. The microRNA in ventricular remodeling: The miR-30 family. Biosci. Rep. 2019, 39, BSR20190788. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C. MicroRNAs and Osteoarthritis. Cells 2018, 7, 92. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Chen, J.; Xie, Z.; Huang, K.; Wang, G.; Yang, Y.; Ni, W.; Chen, Z.; Shi, P.; et al. CircSERPINE2 protects against osteoarthritis by targeting miR-1271 and ETS-related gene. Ann. Rheum. Dis. 2019, 78, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, X.; Wen, X.; Zeng, A.; Mao, G.; Lin, R.; Hu, S.; Liao, W.; Zhang, Z. Inhibition of miR-490-5p promotes human adipose-derived stem cells chondrogenesis and protects chondrocytes via the PITPNM1/PI3K/AKT axis. Front. Cell Dev. Biol. 2020, 8, 573221. [Google Scholar] [CrossRef]
- Chen, L.; Yu, L.; Zhang, R.; Zhu, L.; Shen, W. Correlation of microRNA-146a/b with disease risk, biochemical indices, inflammatory cytokines, overall disease severity, and prognosis of sepsis. Medicine 2020, 99, e19754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, R.F.; Zhang, A.N.; Dong, G.X.; Suo, N.; Wu, Z.P.; Liu, Y.M.; Wang, L.T. MiR-204 promotes fracture healing via enhancing cell viability of osteoblasts. Eur. Rev. Med. Pharmacol. Sci. 2018, 22 (Suppl. 1), 29–35. [Google Scholar] [PubMed]
- Ju, C.; Liu, R.; Zhang, Y.; Zhang, F.; Sun, J.; Lv, X.B.; Zhang, Z. Exosomes May Be the Potential New Direction of Research in Osteoarthritis Management. Biomed. Res. Int. 2019, 2019, 7695768. [Google Scholar] [CrossRef]
- Mao, G.; Zhang, Z.; Hu, S.; Zhang, Z.; Chang, Z.; Huang, Z.; Liao, W.; Kang, Y. Exosomes derived from miR-92a-3poverexpressing human mesenchymal stem cells enhance chondrogenesis and suppress cartilage degradation via targeting WNT5A. Stem Cell Res. Ther. 2018, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int. 2018, 2018, 3057624. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.J.; Li, J.; Yang, X.; Du, S.; Ding, J.; Gao, Y.; Zhang, Y.; Yang, K.; Chen, Q. Evidence that miR-146a attenuates aging-and trauma-induced osteoarthritis by inhibiting Notch1, IL-6, and IL-1 mediated catabolism. Aging Cell 2018, 17, e12752. [Google Scholar] [CrossRef] [PubMed]
- Pourakbari, R.; Khodadadi, M.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. The potential of exosomes in the therapy of the cartilage and bone complications; emphasis on osteoarthritis. Life Sci. 2019, 236, 116861. [Google Scholar] [CrossRef]
- Ni, Z.; Zhou, S.; Li, S.; Kuang, L.; Chen, H.; Luo, X.; Ouyang, J.; He, M.; Du, X.; Chen, L. Exosomes: Roles and therapeutic potential in osteoarthritis. Bone Res. 2020, 8, 1–18. [Google Scholar] [CrossRef]
- Zhan, D.; Cross, A.; Wright, H.L.; Moots, R.J.; Edwards, S.W.; Honsawek, S. Internalization of Neutrophil-Derived Microvesicles Modulates TNFα-Stimulated Proinflammatory Cytokine Production in Human Fibroblast-Like Synoviocytes. Int. J. Mol. Sci. 2021, 22, 7409. [Google Scholar] [CrossRef]
- Lotz, M.K. New developments in osteoarthritis. Posttraumatic osteoarthritis: Pathogenesis and pharmacological treatment options. Arthritis Res. Ther. 2010, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Vesela, B.; Zapletalova, M.; Svandova, E.; Ramesova, A.; Doubek, J.; Lesot, H.; Matalova, E. General Caspase Inhibition in Primary Chondrogenic Cultures Impacts Their Transcription Profile Including Osteoarthritis-Related Factors. Cartilage 2021, 13 (Suppl. 2), 1144S–1154S. [Google Scholar] [CrossRef]
- Katz, J.N.; Arant, K.R.; Loeser, R.F. Diagnosis and Treatment of Hip and Knee Osteoarthritis. JAMA 2021, 325, 568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Tong, P.; Xia, L.; Jin, H. Progress on the early diagnosis of knee osteoarthritis. Zhongguo Gu Shang 2016, 29, 288–291. [Google Scholar] [PubMed]
- Munjal, A.; Bapat, S.; Hubbard, D.; Hunter, M.; Kolhe, R.; Fulzele, S. Advances in Molecular biomarker for early diagnosis of Osteoarthritis. Biomol. Concepts 2019, 10, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.W.-P.; Schlüter-Brust, K.U.; Eysel, P. The Epidemiology, Etiology, Diagnosis, and Treatment of Osteoarthritis of the Knee. Dtsch. Arztebl. Int. 2010, 107, 152. [Google Scholar] [CrossRef]
- Skou, S.T.; Roos, E.M. Physical therapy for patients with knee and hip osteoarthritis: Supervised, active treatment is current best practice. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 1), 112–117. [Google Scholar]
- Ernst, E. Complementary or alternative therapies for osteoarthritis. Nat. Clin. Pract. Rheumatol. 2006, 2, 74–80. [Google Scholar] [CrossRef]
- Cameron, M.; Chrubasik, S. Oral herbal therapies for treating osteoarthritis. Cochrane Database Syst Rev. 2014, 2016, CD002947. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.S.F.; Khaliq, S.A.; Raza, M.L.; Zehra, A.; Siddiqui, M.U.A.; Ul-Hasan, M.M. A comparative study of the common complaints and secondary complications in patients of osteoarthritis on allopathic, homeopathic and herbal system of medicines. Pak. J. Pharm. Sci. 2021, 34, 457–463. [Google Scholar]
- Fernandes, J.C.; Martel-Pelletier, J.; Pelletier, J.-P. The role of cytokines in osteoarthritis pathophysiology. Biorheology 2002, 39, 237–246. [Google Scholar]
- McColl, S.R.; Paquin, R.; Ménard, C.; Beaulieu, A.D. Human neutrophils produce high levels of the interleukin 1 receptor antagonist in response to granulocyte/macrophage colony-stimulating factor and tumor necrosis factor alpha. J. Exp. Med. 1992, 176, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darabos, N.; Haspl, M.; Moser, C.; Darabos, A.; Bartolek, D.; Groenemeyer, D. Intraarticular application of autologous conditioned serum (ACS) reduces bone tunnel widening after ACL reconstructive surgery in a randomized controlled trial. Knee Surg. Sport Traumatol Arthrosc. 2011, 19 (Suppl. 1), 36–46. [Google Scholar] [CrossRef]
- Chevalier, X.; Giraudeau, B.; Conrozier, T.; Marliere, J.; Kiefer, P.; Goupille, P. Safety study of intraarticular injection of interleukin 1 receptor antagonist in patients with painful knee osteoarthritis: A multicenter study. J. Rheumatol. 2005, 32, 1317–1323. [Google Scholar] [PubMed]
- Conaghan, P.G.; Cook, A.D.; Hamilton, J.A.; Tak, P.P. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat. Rev. Rheumatol. 2019, 15, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.L. IL-1 in osteoarthritis: Time for a critical review of the literature. F1000Research 2019, 8, 934. [Google Scholar] [CrossRef] [PubMed]
- Wessely-Szponder, J.; Michalska, J.; Szponder, T.; Żylińska, B.; Tarczyńska, M.; Szubstarski, M. The Role of Antimicrobial Neutrophil Extract in Modification of the Inflammatory Response During Osteochondral Autograft and Allograft Transplantation in Rabbits. J. Comp. Pathol. 2020, 175, 49–63. [Google Scholar] [CrossRef]
- Chang, M.C.; Chiang, P.F.; Kuo, Y.J.; Peng, C.L.; Chen, K.Y.; Chiang, Y.C. Hyaluronan-Loaded Liposomal Dexamethasone–Diclofenac Nanoparticles for Local Osteoarthritis Treatment. Int. J. Mol. Sci. 2021, 22, 665. [Google Scholar] [CrossRef]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, A.; Volarevic, V. Mesenchymal stem cell-based therapy of osteoarthritis: Current knowledge and future perspectives. Biomed. Pharmacother. 2019, 109, 2318–2326. [Google Scholar] [CrossRef]
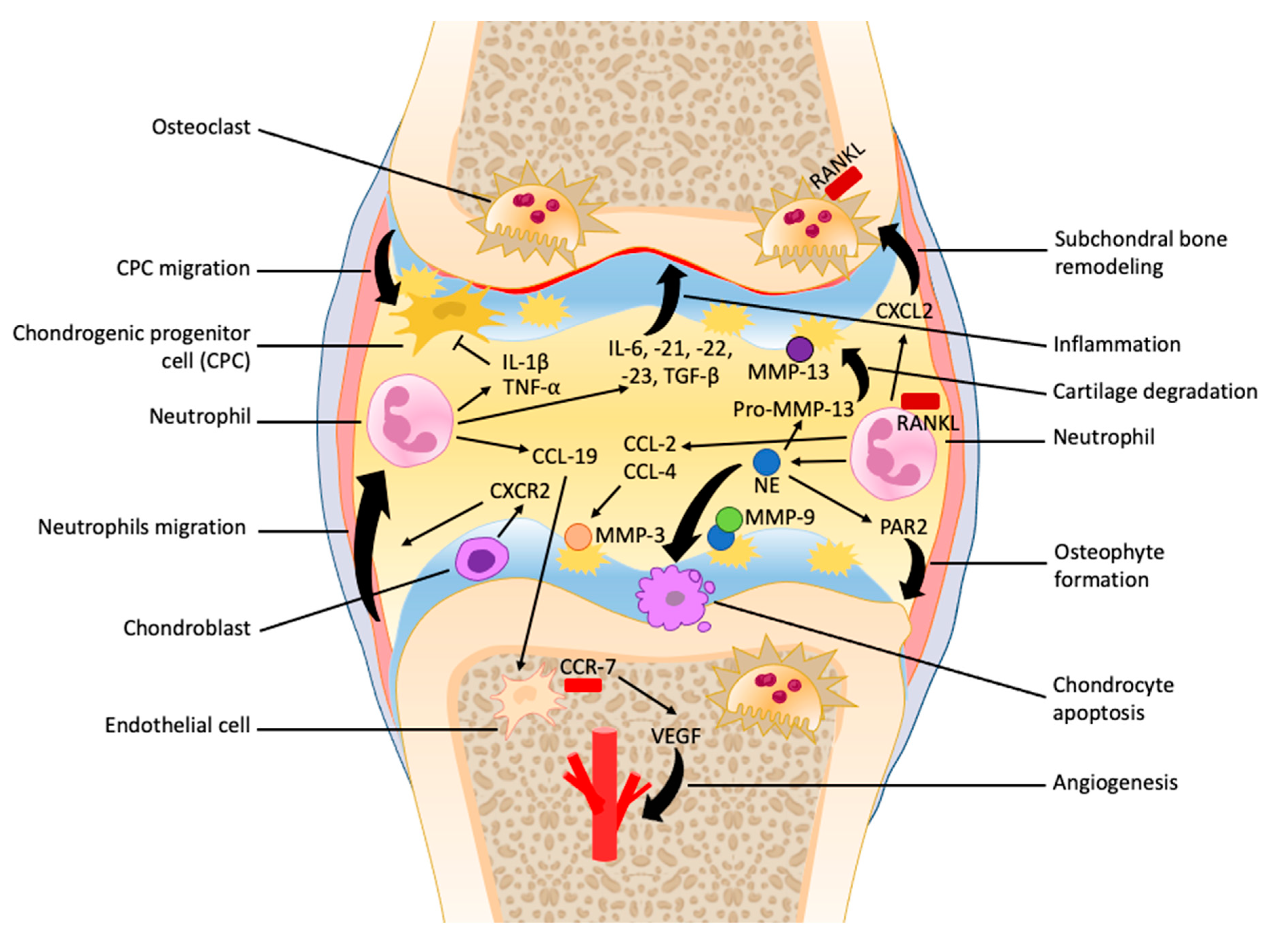

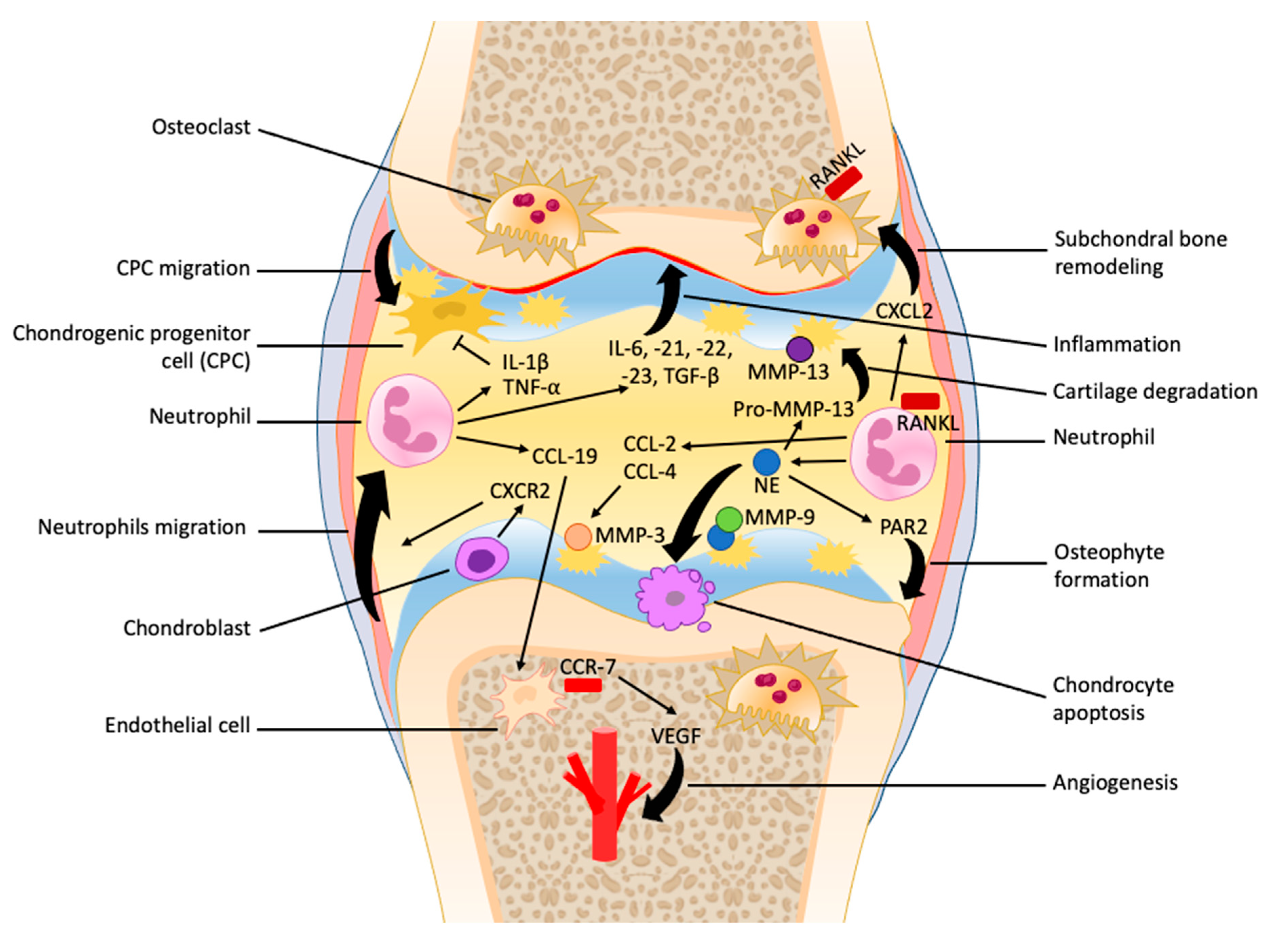{kind=link}
| miRNA | Effects/Interactions | Expression in OA | References |
|---|---|---|---|
| miR-9 | Targets IL-6, NF-ΚB1 and MMP-13 | Downregulated | [48,49] |
| miR-21 | Inhibits GDF5 | Upregulated | [50,51,52] |
| miR-29 | Represses SOX9; targets collagen type 1A1 | Upregulated | [50,51,52] |
| miR-30 | Inhibits autophagy; induces apoptosis | Upregulated | [42,46] |
| miR-36 | Induces expression of inflammatory factors and chemokines via NF-ΚB/A20 signaling | Upregulated | [53] |
| miR-92a | Inhibits histone deacetylase; involved in cartilage development and homeostasis | Downregulated | [50,54,55,56] |
| miR-95 | Involved in cartilage development and homeostasis | Downregulated | [55] |
| miR-99a | Targets FZD1, ITGB5, GDF6; enhances inflammation and apoptosis | Downregulated | [47] |
| miR-100 | Involved in cartilage homeostasis | Downregulated | [55] |
| miR-135b | Involved in chondrocyte proliferation and cartilage repair | Upregulated | [55,57] |
| miR-140 | Induce SOX9, suppress hypertrophy by targeting SMAD1; involved in proliferation and migration of chondrocytes | Downregulated | [50,54,55] |
| miR-141 | Inhibits bone resorption | Upregulated | [39] |
| miR-143 | Targets genes including SMAD3 and DCAKD | Upregulated | [47] |
| miR-146a | Reduces Type II collagen; enhances autophagy and pro-inflammatory cytokines, inhibits NF-ΚB pathway; targets Camk2d | Upregulated | [42,50,58] |
| miR-155 | Inhibits autophagy; overexpression reduces S100A8/19-P secretion of proinflammatory cytokines | Upregulated | [42,46,47] |
| miR-193a | Inhibit inflammation, apoptosis, and cartilage degradation mediators (MMP-3, MMP-13, and ADAMTS)-5; Targets SOX5 | Downregulated | [55] |
| miR-204 | Regulates osteogenesis by targeting Runx2 expression | Downregulated | [50,54] |
| miR-222 | Regulates MMP-13 and histone deacetylase 4; improves chondrogenesis | Downregulated | [50,54] |
| miR-320c | Enhances chondrogenesis by upregulating SOX9 and downregulating MMP-13 and Wnt/β-catenin pathway | Downregulated | [55] |
| miR-370/373 | Inhibits SHMT2 and MECP2 | Downregulated | [50,54] |
| miR-381 | Inhibits histone deacetylase | Downregulated | [50,54] |
| miR-490 | Increases expression of Runx2, and decreases SOX9 | Upregulated | [50,51,52] |
| miR-1271 | Enhances expression of MMP-3, MMP-13 and ADAMTS-4, and decreases levels of SOX9, COL2A1 and aggrecan | Upregulated | [50,51,52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaney, S.; Vergara, R.; Qiryaqoz, Z.; Suggs, K.; Akkouch, A. The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis. Biomedicines 2022, 10, 1604. https://doi.org/10.3390/biomedicines10071604
Chaney S, Vergara R, Qiryaqoz Z, Suggs K, Akkouch A. The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis. Biomedicines. 2022; 10(7):1604. https://doi.org/10.3390/biomedicines10071604
Chicago/Turabian StyleChaney, Shelby, Rosemary Vergara, Zeena Qiryaqoz, Kelsey Suggs, and Adil Akkouch. 2022. "The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis" Biomedicines 10, no. 7: 1604. https://doi.org/10.3390/biomedicines10071604
APA StyleChaney, S., Vergara, R., Qiryaqoz, Z., Suggs, K., & Akkouch, A. (2022). The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis. Biomedicines, 10(7), 1604. https://doi.org/10.3390/biomedicines10071604