
Apolipoprotein A-II, a Player in Multiple Processes and Diseases

 , and
, and
Abstract
1. Introduction
2. ApoA-II Expression and Structural-Functional Properties
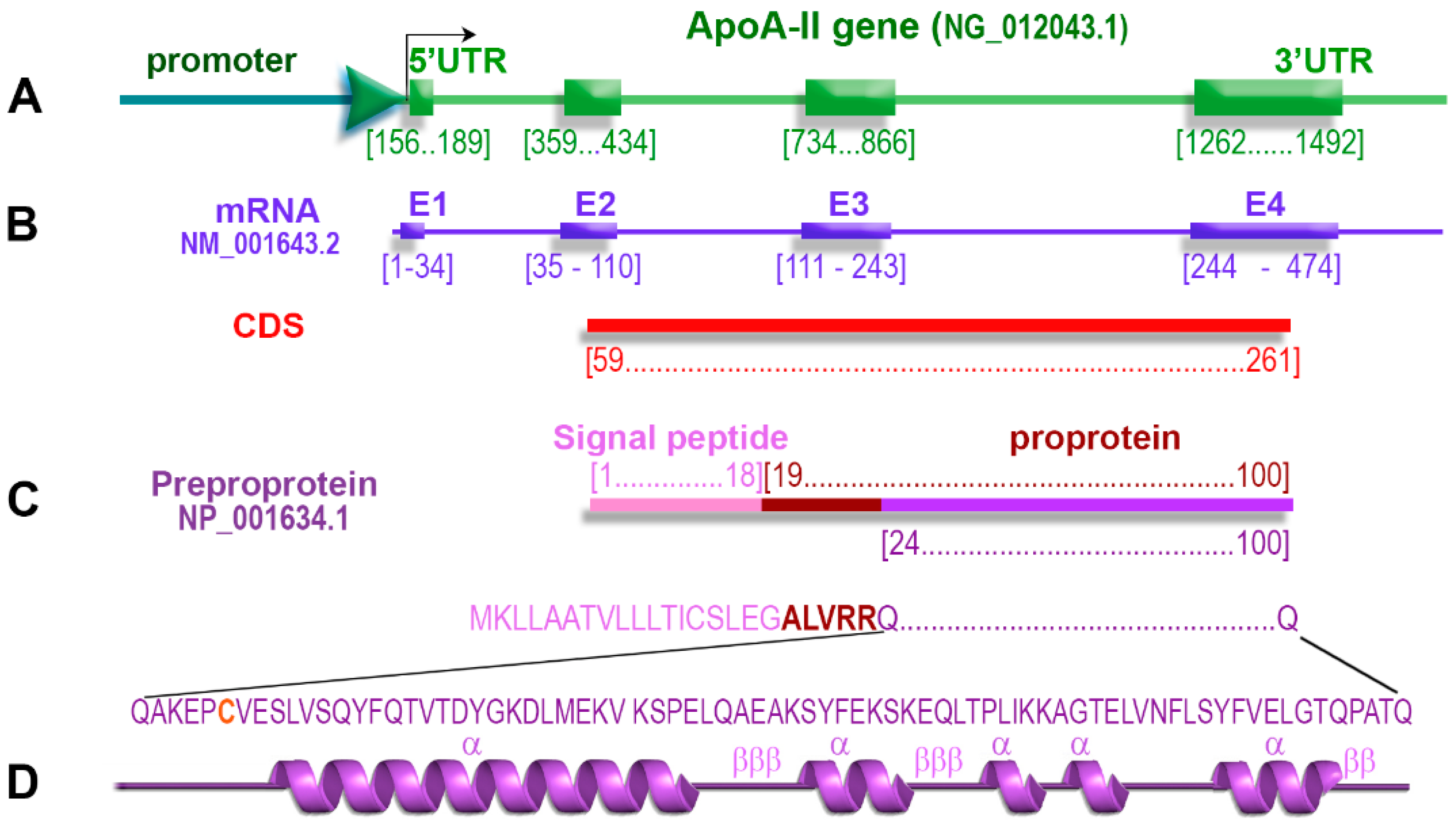
2.1. ApoA-II Gene
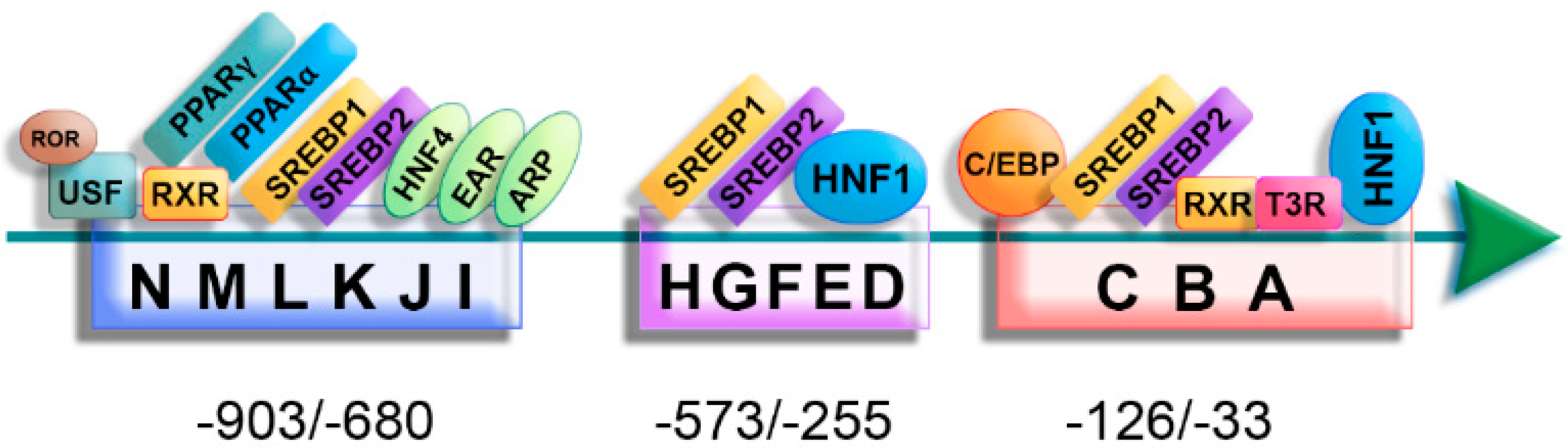
2.2. Regulation of ApoA-II Gene Expression
2.3. The Polymorphisms of the ApoA-II Gene with Clinical Significance
2.4. ApoA-II Protein
2.5. Primary Structure of apoA-II
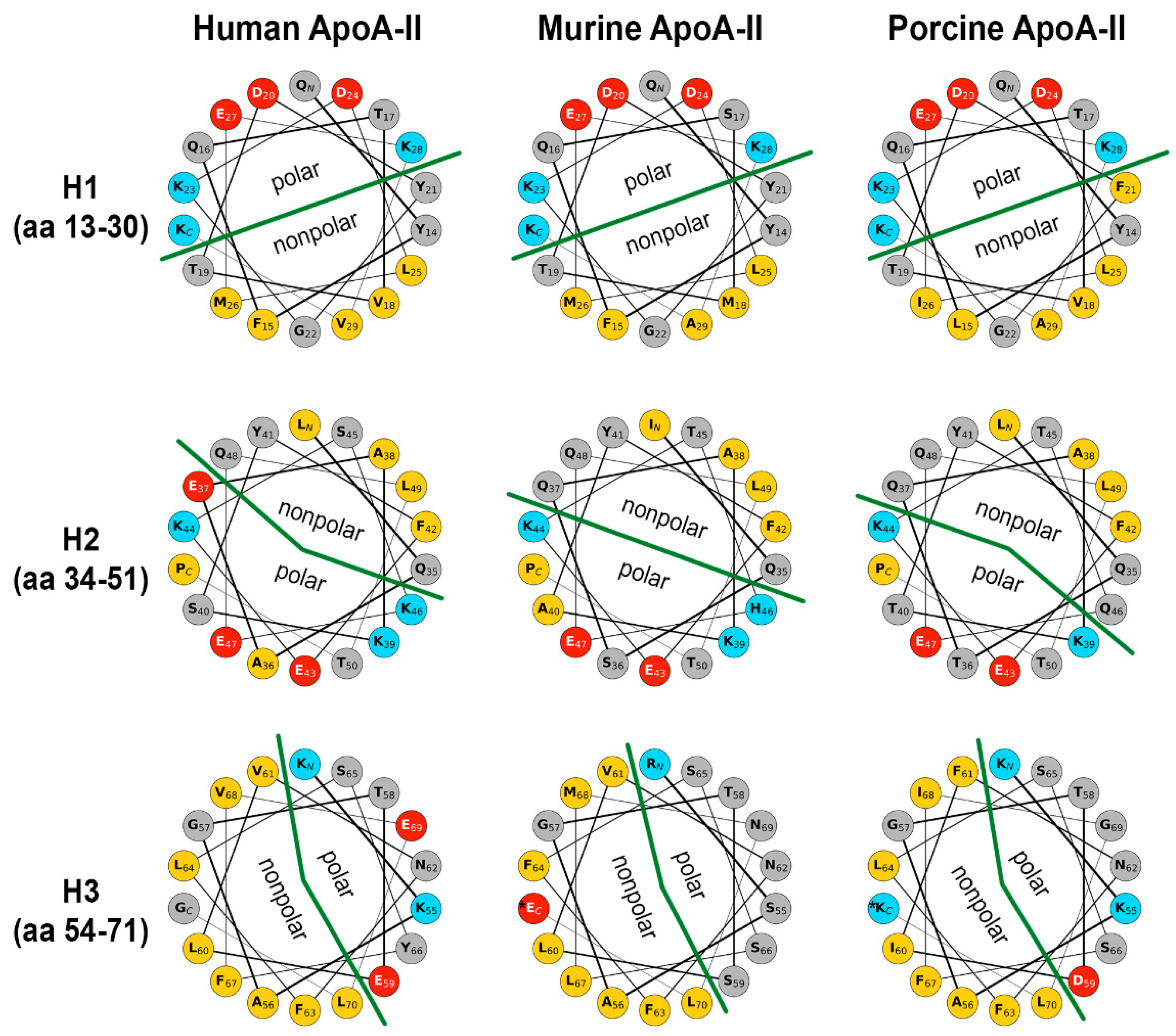
2.6. Secondary Structure of apoA-II
2.7. Quaternary Structure of apoA-II
2.8. Co-Translational Processing of apoA-II
2.9. Post-Translational Modifications of apoA-II
2.10. ApoA-II Isoforms
3. Physiological Roles of apoA-II
4. Role of apoA-II in Various Pathologies
4.1. Role of apoA-II in Atherosclerosis and Cardiovascular Disorders
4.2. Role of apoA-II in Metabolic Syndrome
4.3. Role of apoA-II in Immunity
4.4. Role of apoA-II in the Early Detection of Cancer
4.5. Role of apoA-II in Amyloidosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zannis, V.I.; Fotakis, P.; Koukos, G.; Kardassis, D.; Ehnholm, C.; Jauhiainen, M.; Chroni, A. HDL biogenesis, remodeling, and catabolism. Handb. Exp. Pharmacol. 2015, 224, 53–111. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yuan, S.; Jayaraman, S.; Gursky, O. Role of apolipoprotein A-II in the structure and remodeling of human high-density lipoprotein (HDL): Protein conformational ensemble on HDL. Biochemistry 2012, 51, 4633–4641. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Takata, K.; Peng, R.L.; Kajiyama, G.; Albers, J.J. A splice-junction mutation responsible for familial apolipoprotein A-II deficiency. Am. J. Hum. Genet. 1990, 46, 822–827. [Google Scholar]
- Weng, W.; Breslow, J.L. Dramatically decreased high density lipoprotein cholesterol, increased remnant clearance, and insulin hypersensitivity in apolipoprotein A-II knockout mice suggest a complex role for apolipoprotein A-II in atherosclerosis susceptibility. Proc. Natl. Acad. Sci. USA 1996, 93, 14788–14794. [Google Scholar] [CrossRef] [PubMed]
- Labeur, C.; Lambert, G.; Van Cauteren, T.; Duverger, N.; Vanloo, B.; Chambaz, J.; Vandekerckhove, J.; Castro, G.; Rosseneu, M. Displacement of apo A-I from HDL by apo A-II or its C-terminal helix promotes the formation of pre-beta1 migrating particles and decreases LCAT activation. Atherosclerosis 1998, 139, 351–362. [Google Scholar] [CrossRef]
- Lambert, G.; Decout, A.; Vanloo, B.; Rouy, D.; Duverger, N.; Kalopissis, A.; Vandekerckhove, J.; Chambaz, J.; Brasseur, R.; Rosseneu, M. The C-terminal helix of human apolipoprotein AII promotes the fusion of unilamellar liposomes and displaces apolipoprotein AI from high-density lipoproteins. Eur. J. Biochem. 1998, 253, 328–338. [Google Scholar] [CrossRef]
- Julve, J.; Escola-Gil, J.C.; Rotllan, N.; Fievet, C.; Vallez, E.; de la Torre, C.; Ribas, V.; Sloan, J.H.; Blanco-Vaca, F. Human apolipoprotein A-II determines plasma triglycerides by regulating lipoprotein lipase activity and high-density lipoprotein proteome. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 232–238. [Google Scholar] [CrossRef]
- Boisfer, E.; Stengel, D.; Pastier, D.; Laplaud, P.M.; Dousset, N.; Ninio, E.; Kalopissis, A.D. Antioxidant properties of HDL in transgenic mice overexpressing human apolipoprotein A-II. J. Lipid Res. 2002, 43, 732–741. [Google Scholar] [CrossRef]
- Ribas, V.; Sanchez-Quesada, J.L.; Anton, R.; Camacho, M.; Julve, J.; Escola-Gil, J.C.; Vila, L.; Ordonez-Llanos, J.; Blanco-Vaca, F. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: A new mechanism linking HDL protein composition and antiatherogenic potential. Circ. Res. 2004, 95, 789–797. [Google Scholar] [CrossRef]
- Middleton-Price, H.R.; van den Berghe, J.A.; Scott, J.; Knott, T.J.; Malcolm, S. Regional chromosomal localisation of APOA2 to 1q21–1q23. Hum. Genet. 1988, 79, 283–285. [Google Scholar] [CrossRef]
- Mehrabian, M.; Qiao, J.H.; Hyman, R.; Ruddle, D.; Laughton, C.; Lusis, A.J. Influence of the apoA-II gene locus on HDL levels and fatty streak development in mice. Arterioscler. Thromb. 1993, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chan, L. The apolipoprotein multigene family: Structure, expression, evolution, and molecular genetics. Klin. Wochenschr. 1989, 67, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Ikewaki, K.; Zech, L.A.; Kindt, M.; Brewer, H.B., Jr.; Rader, D.J. Apolipoprotein A-II production rate is a major factor regulating the distribution of apolipoprotein A-I among HDL subclasses LpA-I and LpA-I:A-II in normolipidemic humans. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Cardot, P.; Chambaz, J.; Cladaras, C.; Zannis, V.I. Regulation of the human ApoA-II gene by the synergistic action of factors binding to the proximal and distal regulatory elements. J. Biol. Chem. 1991, 266, 24460–24470. [Google Scholar] [CrossRef]
- Lucero, M.A.; Sanchez, D.; Ochoa, A.R.; Brunel, F.; Cohen, G.N.; Baralle, F.E.; Zakin, M.M. Interaction of DNA-binding proteins with the tissue-specific human apolipoprotein-AII enhancer. Nucleic Acids Res. 1989, 17, 2283–2300. [Google Scholar] [CrossRef]
- Pissios, P.; Kan, H.Y.; Nagaoka, S.; Zannis, V.I. SREBP-1 binds to multiple sites and transactivates the human ApoA-II promoter in vitro: SREBP-1 mutants defective in DNA binding or transcriptional activation repress ApoA-II promoter activity. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1456–1469. [Google Scholar] [CrossRef][Green Version]
- Kan, H.Y.; Pissios, P.; Chambaz, J.; Zannis, V.I. DNA binding specificity and transactivation properties of SREBP-2 bound to multiple sites on the human apoA-II promoter. Nucleic Acids Res. 1999, 27, 1104–1117. [Google Scholar] [CrossRef][Green Version]
- Hatzivassiliou, E.; Koukos, G.; Ribeiro, A.; Zannis, V.; Kardassis, D. Functional specificity of two hormone response elements present on the human apoA-II promoter that bind retinoid X receptor alpha/thyroid receptor beta heterodimers for retinoids and thyroids: Synergistic interactions between thyroid receptor beta and upstream stimulatory factor 2a. Biochem. J. 2003, 376, 423–431. [Google Scholar] [CrossRef][Green Version]
- Cardot, P.; Chambaz, J.; Kardassis, D.; Cladaras, C.; Zannis, V.I. Factors participating in the liver-specific expression of the human apolipoprotein A-II gene and their significance for transcription. Biochemistry 1993, 32, 9080–9093. [Google Scholar] [CrossRef]
- Ladias, J.A.; Hadzopoulou-Cladaras, M.; Kardassis, D.; Cardot, P.; Cheng, J.; Zannis, V.; Cladaras, C. Transcriptional regulation of human apolipoprotein genes ApoB, ApoCIII, and ApoAII by members of the steroid hormone receptor superfamily HNF-4, ARP-1, EAR-2, and EAR-3. J. Biol. Chem. 1992, 267, 15849–15860. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Schoonjans, K.; Kosykh, V.; Dallongeville, J.; Heyman, R.A.; Staels, B.; Auwerx, J. Retinoids increase human apolipoprotein A-11 expression through activation of the retinoid X receptor but not the retinoic acid receptor. Mol. Cell. Biol. 1996, 16, 3350–3360. [Google Scholar] [CrossRef] [PubMed]
- Sauvaget, D.; Chauffeton, V.; Dugue-Pujol, S.; Kalopissis, A.D.; Guillet-Deniau, I.; Foufelle, F.; Chambaz, J.; Leturque, A.; Cardot, P.; Ribeiro, A. In vitro transcriptional induction of the human apolipoprotein A-II gene by glucose. Diabetes 2004, 53, 672–678. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lacorte, J.M.; Beigneux, A.; Parant, M.; Chambaz, J. Repression of apoC-III gene expression by TNFalpha involves C/EBPdelta/NF-IL6beta via an IL-1 independent pathway. FEBS Lett. 1997, 415, 217–220. [Google Scholar] [CrossRef][Green Version]
- Trusca, V.G.; Dumitrescu, M.; Fenyo, I.M.; Tudorache, I.F.; Simionescu, M.; Gafencu, A.V. The Mechanism of Bisphenol A Atherogenicity Involves Apolipoprotein A-I Downregulation through NF-kappaB Activation. Int. J. Mol. Sci. 2019, 20, 6281. [Google Scholar] [CrossRef] [PubMed]
- Gafencu, A.V.; Robciuc, M.R.; Fuior, E.; Zannis, V.I.; Kardassis, D.; Simionescu, M. Inflammatory signaling pathways regulating ApoE gene expression in macrophages. J. Biol. Chem. 2007, 282, 21776–21785. [Google Scholar] [CrossRef] [PubMed]
- Trusca, V.G.; Fuior, E.V.; Kardassis, D.; Simionescu, M.; Gafencu, A.V. The Opposite Effect of c-Jun Transcription Factor on Apolipoprotein E Gene Regulation in Hepatocytes and Macrophages. Int. J. Mol. Sci. 2019, 20, 1471. [Google Scholar] [CrossRef]
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin reduces the endotoxin-induced down-regulation of apolipoprotein E gene expression in macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440. [Google Scholar] [CrossRef]
- Lai, C.Q.; Smith, C.E.; Parnell, L.D.; Lee, Y.C.; Corella, D.; Hopkins, P.; Hidalgo, B.A.; Aslibekyan, S.; Province, M.A.; Absher, D.; et al. Epigenomics and metabolomics reveal the mechanism of the APOA2-saturated fat intake interaction affecting obesity. Am. J. Clin. Nutr. 2018, 108, 188–200. [Google Scholar] [CrossRef]
- Takada, D.; Emi, M.; Ezura, Y.; Nobe, Y.; Kawamura, K.; Iino, Y.; Katayama, Y.; Xin, Y.; Wu, L.L.; Larringa-Shum, S.; et al. Interaction between the LDL-receptor gene bearing a novel mutation and a variant in the apolipoprotein A-II promoter: Molecular study in a 1135-member familial hypercholesterolemia kindred. J. Hum. Genet. 2002, 47, 656–664. [Google Scholar] [CrossRef]
- van‘t Hooft, F.M.; Ruotolo, G.; Boquist, S.; de Faire, U.; Eggertsen, G.; Hamsten, A. Human evidence that the apolipoprotein a-II gene is implicated in visceral fat accumulation and metabolism of triglyceride-rich lipoproteins. Circulation 2001, 104, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lista, J.; Perez-Jimenez, F.; Tanaka, T.; Perez-Martinez, P.; Jimenez-Gomez, Y.; Marin, C.; Ruano, J.; Parnell, L.; Ordovas, J.M.; Lopez-Miranda, J. An apolipoprotein A-II polymorphism (−265T/C, rs5082) regulates postprandial response to a saturated fat overload in healthy men. J. Nutr. 2007, 137, 2024–2028. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, F.; Wiltshire, S.; Hung, J.; Jennens, M.; Beilby, J.P.; Thompson, P.L.; McQuillan, B.M.; McCaskie, P.A.; Carter, K.W.; et al. The apolipoprotein AII rs5082 variant is associated with reduced risk of coronary artery disease in an Australian male population. Atherosclerosis 2008, 199, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Peloso, G.; Arnett, D.K.; Demissie, S.; Cupples, L.A.; Tucker, K.; Lai, C.Q.; Parnell, L.D.; Coltell, O.; Lee, Y.C.; et al. APOA2, dietary fat, and body mass index: Replication of a gene-diet interaction in 3 independent populations. Arch. Intern. Med. 2009, 169, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Tai, E.S.; Sorli, J.V.; Chew, S.K.; Coltell, O.; Sotos-Prieto, M.; Garcia-Rios, A.; Estruch, R.; Ordovas, J.M. Association between the APOA2 promoter polymorphism and body weight in Mediterranean and Asian populations: Replication of a gene-saturated fat interaction. Int. J. Obes. 2011, 35, 666–675. [Google Scholar] [CrossRef]
- Noorshahi, N.; Sotoudeh, G.; Djalali, M.; Eshraghian, M.R.; Keramatipour, M.; Basiri, M.G.; Doostan, F.; Koohdani, F. APOA II genotypes frequency and their interaction with saturated fatty acids consumption on lipid profile of patients with type 2 diabetes. Clin. Nutr. 2016, 35, 907–911. [Google Scholar] [CrossRef]
- Brewer, H.B., Jr.; Lux, S.E.; Ronan, R.; John, K.M. Amino acid sequence of human apoLp-Gln-II (apoA-II), an apolipoprotein isolated from the high-density lipoprotein complex. Proc. Natl. Acad. Sci. USA 1972, 69, 1304–1308. [Google Scholar] [CrossRef]
- Li, W.H.; Tanimura, M.; Luo, C.C.; Datta, S.; Chan, L. The apolipoprotein multigene family: Biosynthesis, structure, structure-function relationships, and evolution. J. Lipid Res. 1988, 29, 245–271. [Google Scholar] [CrossRef]
- Blanco-Vaca, F.; Escola-Gil, J.C.; Martin-Campos, J.M.; Julve, J. Role of apoA-II in lipid metabolism and atherosclerosis: Advances in the study of an enigmatic protein. J. Lipid Res. 2001, 42, 1727–1739. [Google Scholar] [CrossRef]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C., Jr.; Weisgraber, K.H. Plasma lipoproteins: Apolipoprotein structure and function. J. Lipid Res. 1984, 25, 1277–1294. [Google Scholar] [CrossRef]
- Birjmohun, R.S.; Dallinga-Thie, G.M.; Kuivenhoven, J.A.; Stroes, E.S.; Otvos, J.D.; Wareham, N.J.; Luben, R.; Kastelein, J.J.; Khaw, K.T.; Boekholdt, S.M. Apolipoprotein A-II is inversely associated with risk of future coronary artery disease. Circulation 2007, 116, 2029–2035. [Google Scholar] [CrossRef]
- Weisgraber, K.H.; Mahley, R.W. Apoprotein (E--A-II) complex of human plasma lipoproteins. I. Characterization of this mixed disulfide and its identification in a high density lipoprotein subfraction. J. Biol. Chem. 1978, 253, 6281–6288. [Google Scholar] [CrossRef]
- Blanco-Vaca, F.; Via, D.P.; Yang, C.Y.; Massey, J.B.; Pownall, H.J. Characterization of disulfide-linked heterodimers containing apolipoprotein D in human plasma lipoproteins. J. Lipid Res. 1992, 33, 1785–1796. [Google Scholar] [CrossRef]
- Tailleux, A.; Duriez, P.; Fruchart, J.C.; Clavey, V. Apolipoprotein A-II, HDL metabolism and atherosclerosis. Atherosclerosis 2002, 164, 1–13. [Google Scholar] [CrossRef]
- Benetollo, C.; Lambert, G.; Talussot, C.; Vanloo, E.; Cauteren, T.V.; Rouy, D.; Dubois, H.; Baert, J.; Kalopissis, A.; Denefle, P.; et al. Lipid-binding properties of synthetic peptide fragments of human apolipoprotein A-II. Eur. J. Biochem. 1996, 242, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Zhao, Z.; Vanloo, B.; Vos, R.; Deridder, E.; Dhoest, A.; Taveirne, J.; Brouwers, E.; Demarsin, E.; Engelborghs, Y.; et al. Phospholipid binding and lecithin-cholesterol acyltransferase activation properties of apolipoprotein A-I mutants. Biochemistry 1995, 34, 13334–13342. [Google Scholar] [CrossRef]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Murley, Y.M.; Yon, E.; Gillotte, K.L.; Davidson, W.S. Comparison of the structural and functional effects of monomeric and dimeric human apolipoprotein A-II in high density lipoprotein particles. Lipids 1996, 31, 1107–1113. [Google Scholar] [CrossRef]
- Nedelkov, D. Mass Spectrometric Studies of Apolipoprotein Proteoforms and Their Role in Lipid Metabolism and Type 2 Diabetes. Proteomes 2017, 5, 27. [Google Scholar] [CrossRef]
- Chan, D.C.; Ng, T.W.; Watts, G.F. Apolipoprotein A-II: Evaluating its significance in dyslipidaemia, insulin resistance, and atherosclerosis. Ann. Med. 2012, 44, 313–324. [Google Scholar] [CrossRef]
- Gursky, O. Structural stability and functional remodeling of high-density lipoproteins. FEBS Lett. 2015, 589, 2627–2639. [Google Scholar] [CrossRef]
- Halim, A.; Ruetschi, U.; Larson, G.; Nilsson, J. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J. Proteome Res. 2013, 12, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef]
- Pankhurst, G.; Wang, X.L.; Wilcken, D.E.; Baernthaler, G.; Panzenbock, U.; Raftery, M.; Stocker, R. Characterization of specifically oxidized apolipoproteins in mildly oxidized high density lipoprotein. J. Lipid Res. 2003, 44, 349–355. [Google Scholar] [CrossRef]
- Knott, T.J.; Wallis, S.C.; Robertson, M.E.; Priestley, L.M.; Urdea, M.; Rall, L.B.; Scott, J. The human apolipoprotein AII gene: Structural organization and sites of expression. Nucleic Acids Res. 1985, 13, 6387–6398. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Remaley, A.T.; Wong, A.W.; Schumacher, U.K.; Meng, M.S.; Brewer, H.B., Jr.; Hoeg, J.M. O-linked glycosylation modifies the association of apolipoprotein A-II to high density lipoproteins. J. Biol. Chem. 1993, 268, 6785. [Google Scholar] [CrossRef]
- Honda, K.; Okusaka, T.; Felix, K.; Nakamori, S.; Sata, N.; Nagai, H.; Ioka, T.; Tsuchida, A.; Shimahara, T.; Shimahara, M.; et al. Altered plasma apolipoprotein modifications in patients with pancreatic cancer: Protein characterization and multi-institutional validation. PLoS ONE 2012, 7, e46908. [Google Scholar] [CrossRef]
- Honda, K.; Srivastava, S. Potential usefulness of apolipoprotein A2 isoforms for screening and risk stratification of pancreatic cancer. Biomark. Med. 2016, 10, 1197–1207. [Google Scholar] [CrossRef]
- Kobayashi, T.; Sato, Y.; Nishiumi, S.; Yagi, Y.; Sakai, A.; Shiomi, H.; Masuda, A.; Okaya, S.; Kutsumi, H.; Yoshida, M.; et al. Serum apolipoprotein A2 isoforms in autoimmune pancreatitis. Biochem. Biophys. Res. Commun. 2018, 497, 903–907. [Google Scholar] [CrossRef]
- Kobayashi, T.; Honda, K. Trends in biomarker discoveries for the early detection and risk stratification of pancreatic cancer using omics studies. Expert Rev. Mol. Diagn. 2019, 19, 651–654. [Google Scholar] [CrossRef]
- Honda, K. Risk stratification of pancreatic cancer by a blood test for apolipoprotein A2-isoforms. Cancer Biomark. 2022, 33, 503–512. [Google Scholar] [CrossRef]
- Azizkhanian, I.; Trenchevska, O.; Bashawri, Y.; Hu, J.; Koska, J.; Reaven, P.D.; Nelson, R.W.; Nedelkov, D.; Yassine, H.N. Posttranslational modifications of apolipoprotein A-II proteoforms in type 2 diabetes. J. Clin. Lipidol. 2016, 10, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, J.T.; Seckler, H.S.; Rink, J.; Compton, P.D.; Fornelli, L.; Thaxton, C.S.; LeDuc, R.; Jacobs, D.; Doubleday, P.F.; Sniderman, A.; et al. Spectrum of Apolipoprotein AI and Apolipoprotein AII Proteoforms and Their Associations With Indices of Cardiometabolic Health: The CARDIA Study. J. Am. Heart Assoc. 2021, 10, e019890. [Google Scholar] [CrossRef]
- Kihara, T.; Yamagishi, K.; Honda, K.; Ikeda, A.; Yatsuya, H.; Saito, I.; Kokubo, Y.; Yamaji, T.; Shimazu, T.; Sawada, N.; et al. Apolipoprotein A2 Isoforms in Relation to the Risk of Myocardial Infarction: A Nested Case-Control Analysis in the JPHC Study. J. Atheroscler. Thromb. 2021, 28, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C.; Castellani, L.W.; Warden, C.H.; Puppione, D.L.; Lusis, A.J. Influence of mouse apolipoprotein A-II on plasma lipoproteins in transgenic mice. J. Biol. Chem. 1993, 268, 20676–20682. [Google Scholar] [CrossRef]
- Schultz, J.R.; Gong, E.L.; McCall, M.R.; Nichols, A.V.; Clift, S.M.; Rubin, E.M. Expression of human apolipoprotein A-II and its effect on high density lipoproteins in transgenic mice. J. Biol. Chem. 1992, 267, 21630–21636. [Google Scholar] [CrossRef]
- Castellani, L.W.; Navab, M.; Van Lenten, B.J.; Hedrick, C.C.; Hama, S.Y.; Goto, A.M.; Fogelman, A.M.; Lusis, A.J. Overexpression of apolipoprotein AII in transgenic mice converts high density lipoproteins to proinflammatory particles. J. Clin. Investig. 1997, 100, 464–474. [Google Scholar] [CrossRef]
- Kido, T.; Kurata, H.; Kondo, K.; Itakura, H.; Okazaki, M.; Urata, T.; Yokoyama, S. Bioinformatic Analysis of Plasma Apolipoproteins A-I and A-II Revealed Unique Features of A-I/A-II HDL Particles in Human Plasma. Sci. Rep. 2016, 6, 31532. [Google Scholar] [CrossRef]
- Melchior, J.T.; Street, S.E.; Andraski, A.B.; Furtado, J.D.; Sacks, F.M.; Shute, R.L.; Greve, E.I.; Swertfeger, D.K.; Li, H.; Shah, A.S.; et al. Apolipoprotein A-II alters the proteome of human lipoproteins and enhances cholesterol efflux from ABCA1. J. Lipid Res. 2017, 58, 1374–1385. [Google Scholar] [CrossRef]
- Lagocki, P.A.; Scanu, A.M. In vitro modulation of the apolipoprotein composition of high density lipoprotein. Displacement of apolipoprotein A-I from high density lipoprotein by apolipoprotein A-II. J. Biol. Chem. 1980, 255, 3701–3706. [Google Scholar] [CrossRef]
- Zvintzou, E.; Xepapadaki, E.; Kalogeropoulou, C.; Filou, S.; Kypreos, K.E. Pleiotropic effects of apolipoprotein A- on high-density lipoprotein functionality, adipose tissue metabolic activity and plasma glucose homeostasis. J. Biomed. Res. 2019, 34, 14–26. [Google Scholar] [CrossRef]
- Castellani, L.W.; Goto, A.M.; Lusis, A.J. Studies with apolipoprotein A-II transgenic mice indicate a role for HDLs in adiposity and insulin resistance. Diabetes 2001, 50, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Kluth, O.; Matzke, D.; Kamitz, A.; Jahnert, M.; Vogel, H.; Scherneck, S.; Schulze, M.; Staiger, H.; Machicao, F.; Haring, H.U.; et al. Identification of Four Mouse Diabetes Candidate Genes Altering beta-Cell Proliferation. PLoS Genet. 2015, 11, e1005506. [Google Scholar] [CrossRef] [PubMed]
- Manduteanu, I.; Simionescu, M. Inflammation in atherosclerosis: A cause or a result of vascular disorders? J. Cell. Mol. Med. 2012, 16, 1978–1990. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Verstuyft, J.G.; Gong, E.L.; Nichols, A.V.; Rubin, E.M. Protein composition determines the anti-atherogenic properties of HDL in transgenic mice. Nature 1993, 365, 762–764. [Google Scholar] [CrossRef]
- Xepapadaki, E.; Kalogeropoulou, C.; Zvintzou, E.; Filou, S.; Kypreos, K. High density lipoprotein: The role of apolipoprotein A2. Hell. J. Atheroscler. 2019, 9, 128–137. [Google Scholar]
- Mackness, M.I.; Arrol, S.; Durrington, P.N. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett. 1991, 286, 152–154. [Google Scholar] [CrossRef]
- Watson, A.D.; Navab, M.; Hama, S.Y.; Sevanian, A.; Prescott, S.M.; Stafforini, D.M.; McIntyre, T.M.; Du, B.N.; Fogelman, A.M.; Berliner, J.A. Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J. Clin. Investig. 1995, 95, 774–782. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Koike, T.; Kitajima, S.; Yu, Y.; Li, Y.; Nishijima, K.; Liu, E.; Sun, H.; Waqar, A.B.; Shibata, N.; Inoue, T.; et al. Expression of human apoAII in transgenic rabbits leads to dyslipidemia: A new model for combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2047–2053. [Google Scholar] [CrossRef]
- Shimano, H. ApoAII controversy still in rabbit? Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1984–1985. [Google Scholar] [CrossRef]
- Wang, Y.; Niimi, M.; Nishijima, K.; Waqar, A.B.; Yu, Y.; Koike, T.; Kitajima, S.; Liu, E.; Inoue, T.; Kohashi, M.; et al. Human apolipoprotein A-II protects against diet-induced atherosclerosis in transgenic rabbits. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Koike, Y.; Yang, D.; Guo, Y.; Rom, O.; Song, J.; Xu, J.; Chen, Y.; Wang, Y.; Zhu, T.; et al. Human apolipoprotein A-II reduces atherosclerosis in knock-in rabbits. Atherosclerosis 2021, 316, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Castellani, L.W.; Gargalovic, P.; Febbraio, M.; Charugundla, S.; Jien, M.L.; Lusis, A.J. Mechanisms mediating insulin resistance in transgenic mice overexpressing mouse apolipoprotein A-II. J. Lipid Res. 2004, 45, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.H.; Hedrick, C.C.; Qiao, J.H.; Castellani, L.W.; Lusis, A.J. Atherosclerosis in transgenic mice overexpressing apolipoprotein A-II. Science 1993, 261, 469–472. [Google Scholar] [CrossRef]
- Khoo, J.C.; Miller, E.; McLoughlin, P.; Steinberg, D. Prevention of low density lipoprotein aggregation by high density lipoprotein or apolipoprotein A-I. J. Lipid Res. 1990, 31, 645–652. [Google Scholar] [CrossRef]
- Boisfer, E.; Lambert, G.; Atger, V.; Tran, N.Q.; Pastier, D.; Benetollo, C.; Trottier, J.F.; Beaucamps, I.; Antonucci, M.; Laplaud, M.; et al. Overexpression of human apolipoprotein A-II in mice induces hypertriglyceridemia due to defective very low density lipoprotein hydrolysis. J. Biol. Chem. 1999, 274, 11564–11572. [Google Scholar] [CrossRef]
- Allayee, H.; Castellani, L.W.; Cantor, R.M.; de Bruin, T.W.; Lusis, A.J. Biochemical and genetic association of plasma apolipoprotein A-II levels with familial combined hyperlipidemia. Circ. Res. 2003, 92, 1262–1267. [Google Scholar] [CrossRef]
- Maiga, S.F.; Kalopissis, A.D.; Chabert, M. Apolipoprotein A-II is a key regulatory factor of HDL metabolism as appears from studies with transgenic animals and clinical outcomes. Biochimie 2014, 96, 56–66. [Google Scholar] [CrossRef]
- Robins, S.J. Targeting low high-density lipoprotein cholesterol for therapy: Lessons from the Veterans Affairs High-density Lipoprotein Intervention Trial. Am. J. Cardiol. 2001, 88, 19–23. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Duschek, N.; Stojakovic, T.; Ghai, S.; Strassegger, J.; Basic, J.; Scharnagl, H.; Falkensammer, J.; Huber, K.; Assadian, A. Ratio of Apolipoprotein A-II/B Improves Risk Prediction of Postoperative Survival After Carotid Endarterectomy. Stroke 2015, 46, 1700–1703. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kido, T.; Kondo, K.; Kurata, H.; Fujiwara, Y.; Urata, T.; Itakura, H.; Yokoyama, S. ApoA-I/A-II-HDL positively associates with apoB-lipoproteins as a potential atherogenic indicator. Lipids Health Dis. 2017, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Boiko, A.S.; Mednova, I.A.; Kornetova, E.G.; Semke, A.V.; Bokhan, N.A.; Loonen, A.J.M.; Ivanova, S.A. Apolipoprotein serum levels related to metabolic syndrome in patients with schizophrenia. Heliyon 2019, 5, e02033. [Google Scholar] [CrossRef] [PubMed]
- Escola-Gil, J.C.; Julve, J.; Marzal-Casacuberta, A.; Ordonez-Llanos, J.; Gonzalez-Sastre, F.; Blanco-Vaca, F. Expression of human apolipoprotein A-II in apolipoprotein E-deficient mice induces features of familial combined hyperlipidemia. J. Lipid Res. 2000, 41, 1328–1338. [Google Scholar] [CrossRef]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef]
- Kheniser, K.G.; Osme, A.; Kim, C.; Ilchenko, S.; Kasumov, T.; Kashyap, S.R. Temporal Dynamics of High-Density Lipoprotein Proteome in Diet-Controlled Subjects with Type 2 Diabetes. Biomolecules 2020, 10, 520. [Google Scholar] [CrossRef]
- Zaki, M.E.; Amr, K.S.; Abdel-Hamid, M. APOA2 Polymorphism in Relation to Obesity and Lipid Metabolism. Cholesterol 2013, 2013, 289481. [Google Scholar] [CrossRef]
- Lara-Castro, C.; Hunter, G.R.; Lovejoy, J.C.; Gower, B.A.; Fernandez, J.R. Apolipoprotein A-II polymorphism and visceral adiposity in African-American and white women. Obes. Res. 2005, 13, 507–512. [Google Scholar] [CrossRef]
- Zaki, M.E.; Amr, K.S.; Abdel-Hamid, M. Evaluating the association of APOA2 polymorphism with insulin resistance in adolescents. Meta Gene 2014, 2, 366–373. [Google Scholar] [CrossRef]
- Furlaneto, C.J.; Ribeiro, F.P.; Hatanaka, E.; Souza, G.M.; Cassatella, M.A.; Campa, A. Apolipoproteins A-I and A-II downregulate neutrophil functions. Lipids 2002, 37, 925–928. [Google Scholar] [CrossRef]
- Thompson, P.A.; Berbee, J.F.; Rensen, P.C.; Kitchens, R.L. Apolipoprotein A-II augments monocyte responses to LPS by suppressing the inhibitory activity of LPS-binding protein. Innate Immun. 2008, 14, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Berbee, J.F.; Havekes, L.M.; Rensen, P.C. Apolipoproteins modulate the inflammatory response to lipopolysaccharide. J. Endotoxin Res. 2005, 11, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed]
- Wait, R.; Chiesa, G.; Parolini, C.; Miller, I.; Begum, S.; Brambilla, D.; Galluccio, L.; Ballerio, R.; Eberini, I.; Gianazza, E. Reference maps of mouse serum acute-phase proteins: Changes with LPS-induced inflammation and apolipoprotein A-I and A-II transgenes. Proteomics 2005, 5, 4245–4253. [Google Scholar] [CrossRef]
- Bagdade, J.D.; Jilma, B.; Hudgins, L.C.; Alaupovic, P.; McCurdy, C.E. LpA-II:B:C:D:E: A new immunochemically-defined acute phase lipoprotein in humans. Lipids Health Dis. 2018, 17, 127. [Google Scholar] [CrossRef]
- Yamashita, J.; Iwamura, C.; Sasaki, T.; Mitsumori, K.; Ohshima, K.; Hada, K.; Hara, N.; Takahashi, M.; Kaneshiro, Y.; Tanaka, H.; et al. Apolipoprotein A-II suppressed concanavalin A-induced hepatitis via the inhibition of CD4 T cell function. J. Immunol. 2011, 186, 3410–3420. [Google Scholar] [CrossRef]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia is associated with the severity of COVID-19. J. Clin. Lipidol. 2020, 14, 297–304. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Zhao, X.; Dong, H.; Wu, C.; Wu, F.; Yu, B.; Lv, J.; Zhang, S.; Wu, G.; et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: An observational study. Lipids Health Dis. 2020, 19, 204. [Google Scholar] [CrossRef]
- Lassale, C.; Hamer, M.; Hernaez, A.; Gale, C.R.; Batty, G.D. Association of pre-pandemic high-density lipoprotein cholesterol with risk of COVID-19 hospitalisation and death: The UK Biobank cohort study. medRxiv 2021. [Google Scholar] [CrossRef]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Velia, E.; Mavingui, P.; et al. Altered high-density lipoprotein composition and functions during severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, J.R.; Lee, I.C.; Kwon, H.J. Native High-Density Lipoproteins (HDL) with Higher Paraoxonase Exerts a Potent Antiviral Effect against SARS-CoV-2 (COVID-19), While Glycated HDL Lost the Antiviral Activity. Antioxidants 2021, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H. Importance of Apolipoprotein A-I and A-II Composition in HDL and Its Potential for Studying COVID-19 and SARS-CoV-2. Medicines 2021, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Kobayashi, M.; Okusaka, T.; Rinaudo, J.A.; Huang, Y.; Marsh, T.; Sanada, M.; Sasajima, Y.; Nakamori, S.; Shimahara, M.; et al. Plasma biomarker for detection of early stage pancreatic cancer and risk factors for pancreatic malignancy using antibodies for apolipoprotein-AII isoforms. Sci. Rep. 2015, 5, 15921. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, M.; Felix, K.; Hartmann, D.; Schnolzer, M.; Nees, M.; Vorderwulbecke, S.; Bogumil, R.; Buchler, M.W.; Friess, H. Identification of potential markers for the detection of pancreatic cancer through comparative serum protein expression profiling. Pancreas 2007, 34, 205–214. [Google Scholar] [CrossRef]
- Xue, A.; Scarlett, C.J.; Chung, L.; Butturini, G.; Scarpa, A.; Gandy, R.; Wilson, S.R.; Baxter, R.C.; Smith, R.C. Discovery of serum biomarkers for pancreatic adenocarcinoma using proteomic analysis. Br. J. Cancer 2010, 103, 391–400. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Li, W.; Zheng, X.; Liu, J.; Wang, J.; Du, G. Apolipoproteins and cancer. Cancer Med. 2019, 8, 7032–7043. [Google Scholar] [CrossRef]
- Malik, G.; Ward, M.D.; Gupta, S.K.; Trosset, M.W.; Grizzle, W.E.; Adam, B.L.; Diaz, J.I.; Semmes, O.J. Serum levels of an isoform of apolipoprotein A-II as a potential marker for prostate cancer. Clin. Cancer Res. 2005, 11, 1073–1085. [Google Scholar] [CrossRef]
- Julovi, S.M.; Xue, A.; Thanh, L.T.; Gill, A.J.; Bulanadi, J.C.; Patel, M.; Waddington, L.J.; Rye, K.A.; Moghaddam, M.J.; Smith, R.C. Apolipoprotein A-II Plus Lipid Emulsion Enhance Cell Growth via SR-B1 and Target Pancreatic Cancer In Vitro and In Vivo. PLoS ONE 2016, 11, e0151475. [Google Scholar] [CrossRef]
- Muchtar, E.; Dispenzieri, A.; Magen, H.; Grogan, M.; Mauermann, M.; McPhail, E.D.; Kurtin, P.J.; Leung, N.; Buadi, F.K.; Dingli, D.; et al. Systemic amyloidosis from A (AA) to T (ATTR): A review. J. Intern. Med. 2021, 289, 268–292. [Google Scholar] [CrossRef]
- Benson, M.D.; Liepnieks, J.J.; Yazaki, M.; Yamashita, T.; Hamidi Asl, K.; Guenther, B.; Kluve-Beckerman, B. A new human hereditary amyloidosis: The result of a stop-codon mutation in the apolipoprotein AII gene. Genomics 2001, 72, 272–277. [Google Scholar] [CrossRef]
- Yazaki, M.; Liepnieks, J.J.; Yamashita, T.; Guenther, B.; Skinner, M.; Benson, M.D. Renal amyloidosis caused by a novel stop-codon mutation in the apolipoprotein A-II gene. Kidney Int. 2001, 60, 1658–1665. [Google Scholar] [CrossRef]
- Yazaki, M.; Liepnieks, J.J.; Barats, M.S.; Cohen, A.H.; Benson, M.D. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003, 64, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Gursky, O. Hot spots in apolipoprotein A-II misfolding and amyloidosis in mice and men. FEBS Lett. 2014, 588, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Prokaeva, T.; Akar, H.; Spencer, B.; Havasi, A.; Cui, H.; O’Hara, C.J.; Gursky, O.; Leszyk, J.; Steffen, M.; Browning, S.; et al. Hereditary Renal Amyloidosis Associated With a Novel Apolipoprotein A-II Variant. Kidney Int. Rep. 2017, 2, 1223–1232. [Google Scholar] [CrossRef]
- Chabert, M.; Rousset, X.; Colombat, M.; Lacasa, M.; Kakanakou, H.; Bourderioux, M.; Brousset, P.; Burlet-Schiltz, O.; Liepnieks, J.J.; Kluve-Beckerman, B.; et al. A transgenic mouse model reproduces human hereditary systemic amyloidosis. Kidney Int. 2019, 96, 628–641. [Google Scholar] [CrossRef]
- Benson, M.D.; Kalopissis, A.D.; Charbert, M.; Liepnieks, J.J.; Kluve-Beckerman, B. A transgenic mouse model of human systemic ApoA2 amyloidosis. Amyloid 2011, 18 (Suppl. 1), 32–33. [Google Scholar] [CrossRef]
- Luo, H.; Sawashita, J.; Tian, G.; Liu, Y.; Li, L.; Ding, X.; Xu, Z.; Yang, M.; Miyahara, H.; Mori, M.; et al. Extracellular deposition of mouse senile AApoAII amyloid fibrils induced different unfolded protein responses in the liver, kidney, and heart. Lab. Investig. 2015, 95, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Kogishi, K.; Wang, J.; Chen, X.; Chiba, T.; Matsushita, T.; Hoshii, Y.; Kawano, H.; Ishihara, T.; Yokota, T.; et al. Fibrilization in mouse senile amyloidosis is fibril conformation-dependent. Lab. Investig. 1998, 78, 1535–1542. [Google Scholar]
- Korenaga, T.; Fu, X.; Xing, Y.; Matsusita, T.; Kuramoto, K.; Syumiya, S.; Hasegawa, K.; Naiki, H.; Ueno, M.; Ishihara, T.; et al. Tissue distribution, biochemical properties, and transmission of mouse type A AApoAII amyloid fibrils. Am. J. Pathol. 2004, 164, 1597–1606. [Google Scholar] [CrossRef]
- Wang, Y.; Sawashita, J.; Qian, J.; Zhang, B.; Fu, X.; Tian, G.; Chen, L.; Mori, M.; Higuchi, K. ApoA-I deficiency in mice is associated with redistribution of apoA-II and aggravated AApoAII amyloidosis. J. Lipid Res. 2011, 52, 1461–1470. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Dai, J.; Li, L.; Ding, X.; Xu, Z.; Mori, M.; Miyahara, H.; Sawashita, J.; Higuchi, K. Apolipoprotein A-II induces acute-phase response associated AA amyloidosis in mice through conformational changes of plasma lipoprotein structure. Sci. Rep. 2018, 8, 5620. [Google Scholar] [CrossRef]
- Sawashita, J.; Kametani, F.; Hasegawa, K.; Tsutsumi-Yasuhara, S.; Zhang, B.; Yan, J.; Mori, M.; Naiki, H.; Higuchi, K. Amyloid fibrils formed by selective N-, C-terminal sequences of mouse apolipoprotein A-II. Biochim. Biophys. Acta 2009, 1794, 1517–1529. [Google Scholar] [CrossRef]
- Sawashita, J.; Zhang, B.; Hasegawa, K.; Mori, M.; Naiki, H.; Kametani, F.; Higuchi, K. C-terminal sequence of amyloid-resistant type F apolipoprotein A-II inhibits amyloid fibril formation of apolipoprotein A-II in mice. Proc. Natl. Acad. Sci. USA 2015, 112, E836–E845. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Wang, J.; Mastushita, T.; Kogishi, K.; Hosokawa, M.; Fu, X.; Guo, Z.; Mori, M.; Higuchi, K. Polymorphisms of mouse apolipoprotein A-II: Seven alleles found among 41 inbred strains of mice. Amyloid 2003, 10, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Li, Y.; Kametani, F.; Cui, X.; Igarashi, Y.; Huo, J.; Miyahara, H.; Mori, M.; Higuchi, K. Curcumin promotes AApoAII amyloidosis and peroxisome proliferation in mice by activating the PPARalpha signaling pathway. Elife 2021, 10, e63538. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Garcia, D.; Lu, Y.; Ozuna, K.; Adjeroh, D.A.; Wang, K.; on Behalf of the Alzheimer’s Disease Neuroimaging Initiative. Levels of Angiotensin-Converting Enzyme and Apolipoproteins Are Associated with Alzheimer’s Disease and Cardiovascular Diseases. Cells 2021, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Granel, B.; Valleix, S.; Serratrice, J.; Cherin, P.; Texeira, A.; Disdier, P.; Weiller, P.J.; Grateau, G. Lysozyme amyloidosis: Report of 4 cases and a review of the literature. Medicine 2006, 85, 66–73. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Clinical Significance and Condition | Chromosome Position | Variation | Location | References |
|---|---|---|---|---|---|
| rs5082 | Pathogenic SNP: Reduced plasma LDL cholesterol Familial hypercholesterolemia 1 | 161,223,893(-) | G/A | REGULATORY | [28,29,30,31,32,33,34,35] |
| rs771259264 | Pathogenic SNP: ApoA-II deficiency, familial (Hiroshima) | 161,222,917(-) | C/T | SPLICE_ DONOR | [3] |
| Isoform | Associated Pathology | Effect of Amino Acid Change | Reference |
|---|---|---|---|
| -ATQ/-ATQ | Autoimmune pancreatitis | ↑ protein expression | [56] |
| Seemingly healthy subjects | [59] | ||
| Autoimmune pancreatitis | ↓ protein expression | [56] | |
| T2D | ↑ degree of oxidation | [61] | |
| Seemingly healthy subjects | [59] | ||
| -ATQ/-AT | Autoimmune pancreatitis chronic pancreatitis; pancreatic cancer | ↓ protein expression | [59] |
| T2D | ↑ degree of oxidation | [61] | |
| Seemingly healthy subjects | [56,57,59] | ||
| -AT/-A | Pancreatic cancer | [59] | |
| -A/-A | Pancreatic cancer | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florea, G.; Tudorache, I.F.; Fuior, E.V.; Ionita, R.; Dumitrescu, M.; Fenyo, I.M.; Bivol, V.G.; Gafencu, A.V. Apolipoprotein A-II, a Player in Multiple Processes and Diseases. Biomedicines 2022, 10, 1578. https://doi.org/10.3390/biomedicines10071578
Florea G, Tudorache IF, Fuior EV, Ionita R, Dumitrescu M, Fenyo IM, Bivol VG, Gafencu AV. Apolipoprotein A-II, a Player in Multiple Processes and Diseases. Biomedicines. 2022; 10(7):1578. https://doi.org/10.3390/biomedicines10071578
Chicago/Turabian StyleFlorea, Gabriela, Irina Florina Tudorache, Elena Valeria Fuior, Radu Ionita, Madalina Dumitrescu, Ioana Madalina Fenyo, Violeta Georgeta Bivol, and Anca Violeta Gafencu. 2022. "Apolipoprotein A-II, a Player in Multiple Processes and Diseases" Biomedicines 10, no. 7: 1578. https://doi.org/10.3390/biomedicines10071578
APA StyleFlorea, G., Tudorache, I. F., Fuior, E. V., Ionita, R., Dumitrescu, M., Fenyo, I. M., Bivol, V. G., & Gafencu, A. V. (2022). Apolipoprotein A-II, a Player in Multiple Processes and Diseases. Biomedicines, 10(7), 1578. https://doi.org/10.3390/biomedicines10071578