1. Introduction
Protein is the basic component of organisms and participates in almost all biological processes in cells [
1,
2]. The vast majority of life activities are the result of the simultaneous action of many proteins, and the interacting protein system is the basis for all life activities. Exploring the interaction between proteins (PPIs) is not only of great significance to the regulation of cell growth, but also lays a theoretical foundation for deeper disease research [
3,
4,
5,
6]. With the fast growth of high-throughput experimental techniques for measuring the interactions between organisms, massive amounts of experimental data on various types of proteins continue to accumulate. This makes it possible to develop effective new theories of analysis and computation that can contribute to a deeper understanding of the mechanisms through which cellular functions arise, providing useful information for studies such as the discovery of evolutionary patterns and even the pathogenic mechanisms of organisms.
In the interaction between proteins, self-interacting protein (SIP) accounts for a large proportion. SIP is an interaction between the same type of proteins, which holds a crucial position as a coordinator in gene expression and other complex biological functions. In-depth studies can help to better understand cancer, viral infections, neurodevelopmental disorders, and other potential diseases disturbed by these factors [
7,
8,
9,
10]. In their study of the origin of protein evolution, Pereira Leal et al. [
11] discovered that proteins evolved through the replication of homodimers, in other words, through the replication of genes encoding SIPs. Wagner et al. [
12] found, in fly, worm, and yeast data sets, that the PPIs between paralogous dimers and homodimers are not independent, and the frequency of the protein–protein interactions between paralogous proteins are higher than that of pure chance. Thus, they believe that gene differentiation and replication are important factors to promote the eukaryotic proteome expansion, and a multicopy of the same subunit is an economic way to form a larger functional structure. Ispolatov et al. [
13] identified a significant amount of SIP in the PPI network, which suggests that it may play a vital function in the cellular system. Moreover, SIP is able to regulate protein function without increasing the genome size, thus expanding its functional diversity.
With the deepening of computational biology research and the accumulation of experimental data [
14,
15,
16], the computational methods of protein–protein interaction prediction are emerging, and excellent results have been achieved [
10,
17,
18,
19,
20]. Among them, the neural network plays an important role. It has a self-learning ability and associative storage function. It can learn the distribution rule of original biological data and find the optimal solution of a computational problem at high speed, so as to effectively improve the performance of the model. For instance, Wang et al. [
21] designed the CNNFSRF method to accurately predict PPI by extracting the features of the protein sequences through convolutional neural networks. In order to effectively combine the key sequence-pattern and sequence-order information when processing the protein samples, Jia et al. [
22] used chaos game to represent information for predicting PPI. You et al. [
23] designed a hierarchical PPI prediction model, PCAEELM, based on protein sequences only. The model refines the protein data features using the PCA algorithm, and is combined with a high-performance ELM classifier to achieve a high accuracy. Chen et al. [
24] combined the location-specific score matrix with wavelet transform to extract protein features, and effectively predicted SIPs using the deep forest-based predictor. In both human and yeast data sets, the proposed model achieves competitive accuracy. Wang et al. [
25] designed the computing method to predict PPI based on the similarity between protein sequences and natural language, and achieved a better performance in the cross-validation experiments. Liu et al. [
26] proposed an SIP prediction computational approach called SPAR, which considers fine-grained domain interaction information to design an improved coding scheme. Compared with other SIP prediction models, this method shows a better performance.
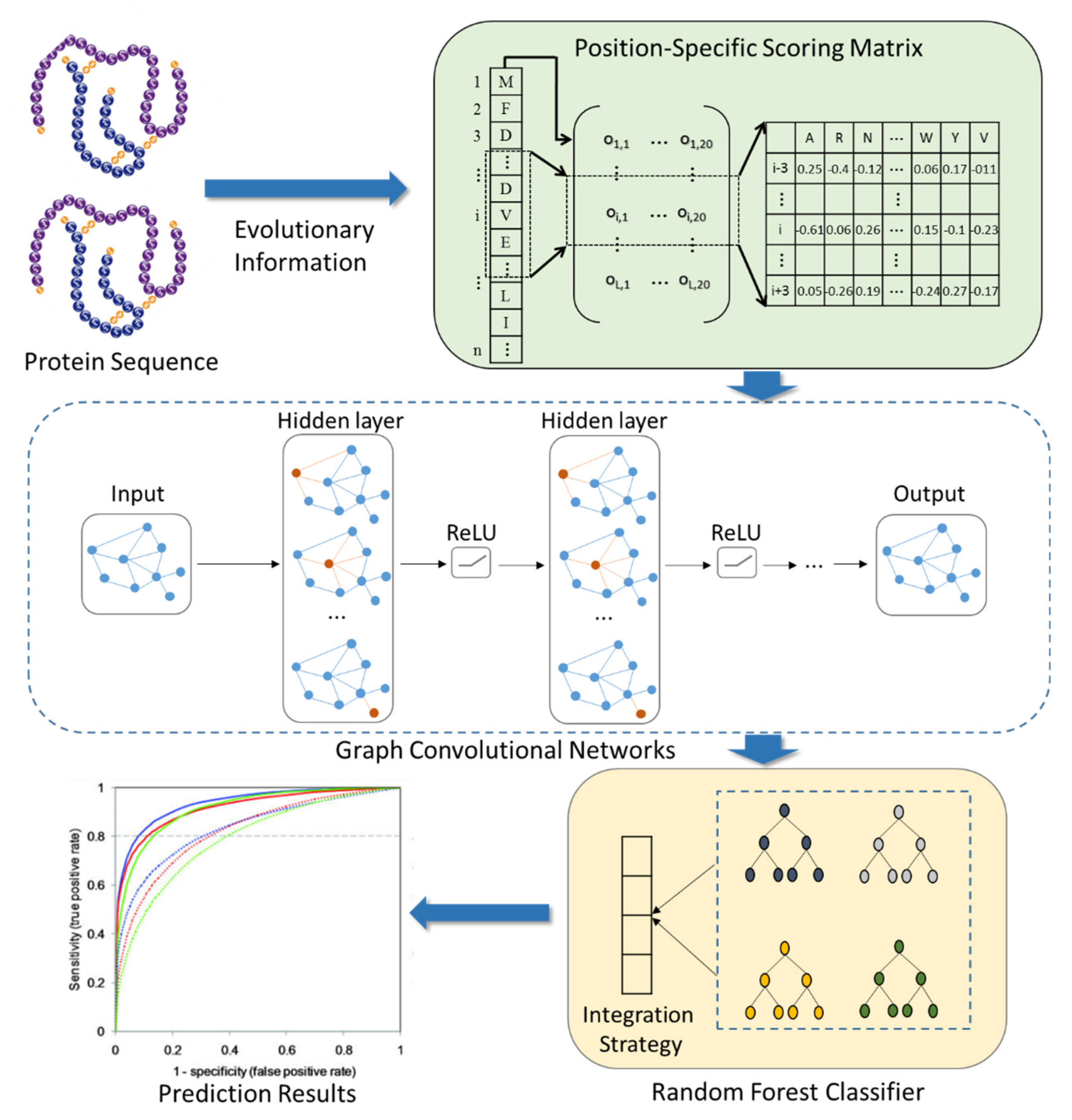
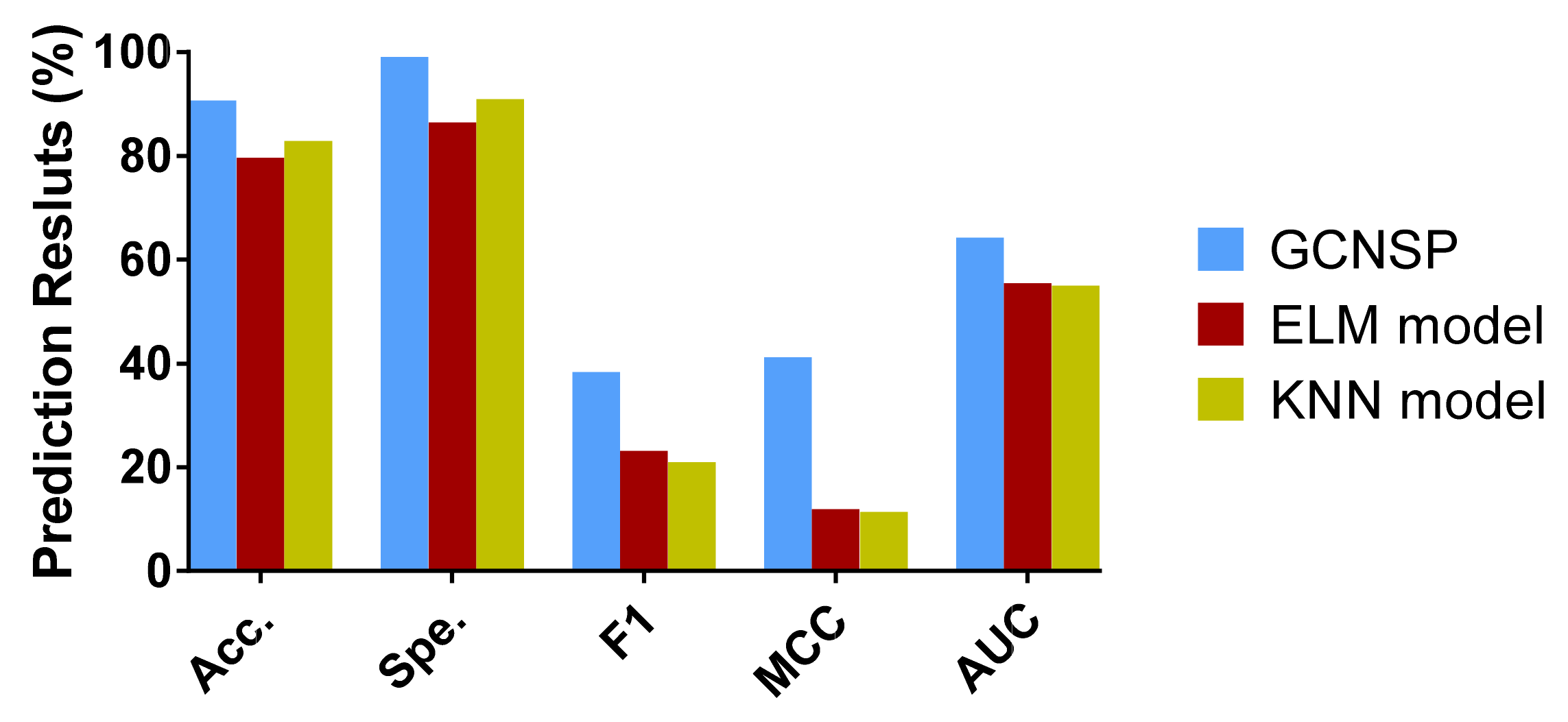
In this work, we combine a deep learning graph convolutional neural network with protein sequences to design the SIP prediction model, SIPGCN. To be specific, SIPGCN first characterizes the biological evolutionary message of the protein amino acid by the PSSM matrix, then extracts their objective distribution features using GCN, and finally predicts the existence of interacting SIPs using an RF classifier. In the human data set, SIPGCN obtained an accuracy of 93.65% and a specificity of 99.64%. In the yeast data set, SIPGCN achieved 90.69% and 99.08% for these evaluation metrics, respectively. Compared with the previous models, SIPGCN exhibited superior competitiveness. The outcomes of these experiments demonstrate that SIPGCN can effectively predict SIPs with interactions from massive quantities of data and can provide reliable candidates for subsequent wet experiments, the flowchart of which is illustrated in
Figure 1. The source code and data used by SIPGCN can be downloaded from
https://github.com/look0012/SIPGCN (accessed on 25 May 2022).
2. Materials and Methods
2.1. Gold Standard Data Sources
In this experiment, we collected two identical proteins from the relevant databases and their interaction mode was described as “direct interaction” to construct the experimental data set. More concretely, human protein sequences with self-interrelation were collated from databases including InnateDB [
27], BioGRID [
28], UniProt [
29], DIP [
30], and MatrixDB [
31]. The principles for selecting these data are as follows: Firstly, the length of protein residues was 50 to 5000 residues. Secondly, only proteins that met one of the following conditions could be selected as positive samples: (1) officially reported by two or more journals, (2) proteins defined as homo-oligomers by the UniProt database, and (3) verified by more than two large-scale or one small-scale experiments. Finally, the negative samples did not contain proteins with self-interaction. Through screening of the above principles, 1441 SIPs and 15,938 non-SIPs were included in the human data set. Additionally, the yeast data set also underwent the same screening, and the quantity of positive and negative samples was 710 and 5511, respectively.
In this study, the data set we used was imbalanced, with the number of negative samples being much larger than the number of positive samples. Generally speaking, the vast majority of data sets in the real world are imbalanced. We would be very lucky if we could obtain a balanced data set. Therefore, to solve the problem of imbalanced data, researchers have put forward many solutions, which can be roughly divided into two categories: one is to build balanced data sets and the other is to use different evaluation indicators to measure the imbalanced data sets.
For the first scheme, we used the resampling method. For example, we used the over-sampling method to increase the number of minority class samples to the same number as that of the majority classes. Another is the use of the undersampling method to select part of the majority class samples to reduce them to the same number as the minority class samples. In addition, the data set can be balanced by generating more virtual samples through GAN and other methods.
For the second scheme, accuracy was not a good measure. It was more inclined to the majority of samples, which is often misleading. Therefore, in addition to accuracy, some other evaluation indicators need to be added to measure the performance of the model. For example, a comprehensive evaluation index F1 that can reflect the accuracy and recall rate, namely, an AUC that can consider the classification ability of the classifier for positive and negative samples at the same time, and can still make a reasonable evaluation of the classifier in the case of unbalanced samples.
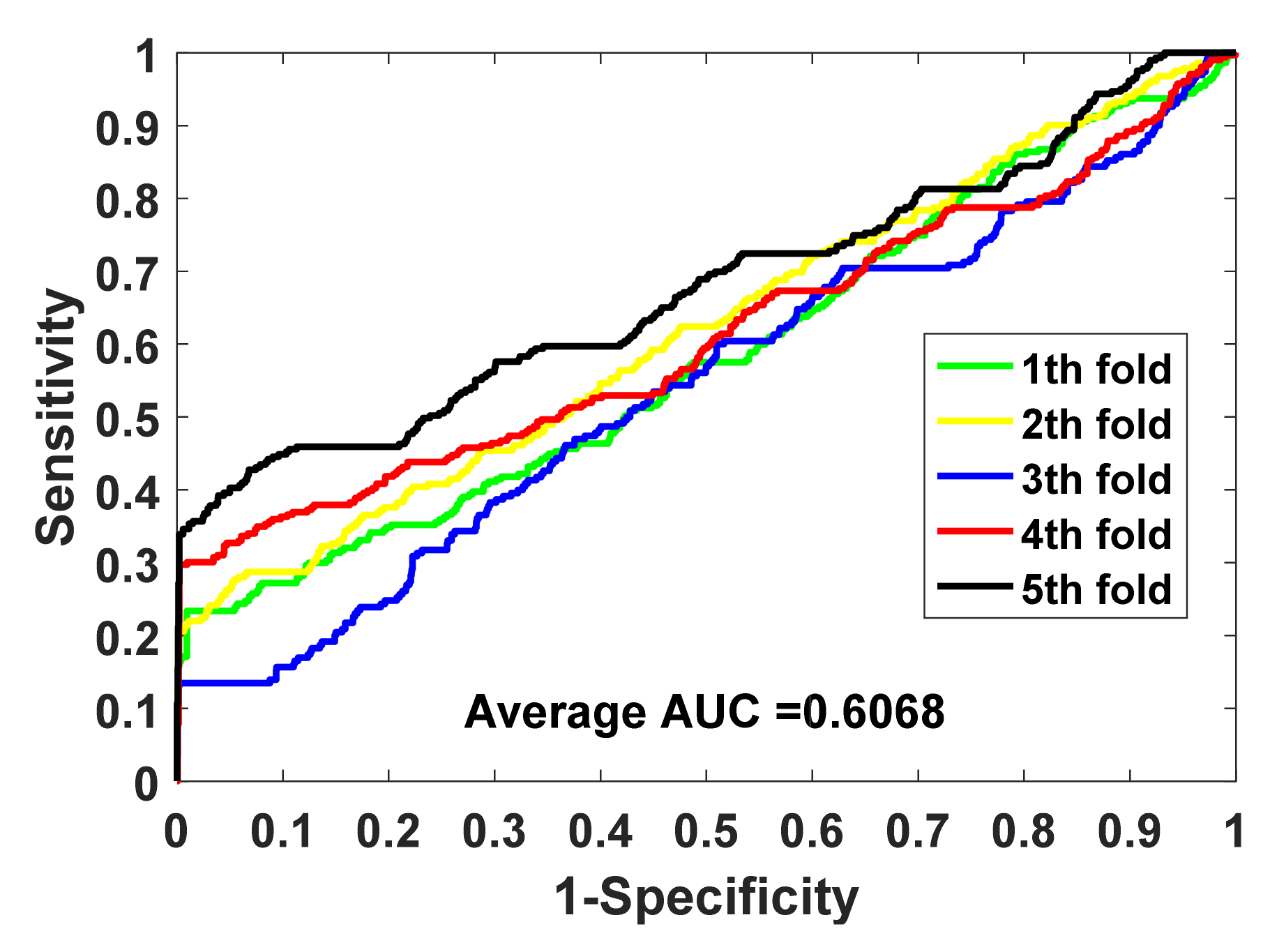
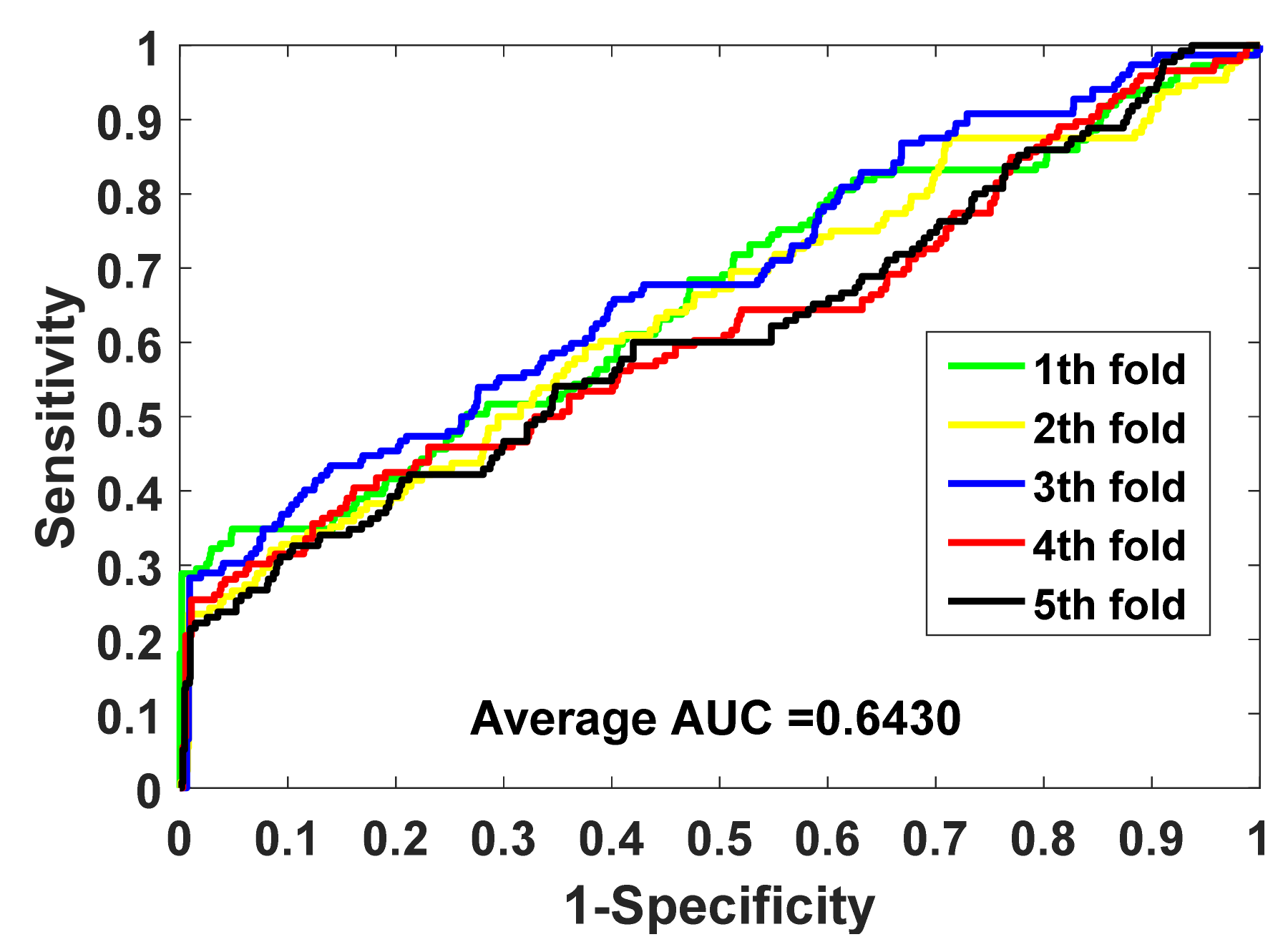
In this study, the gold standard data set we used produced both positive and negative samples. Unlike some data sets that only produced positive samples, we needed to build negative samples (in this case, we built a balanced data set). Based on the consideration of maintaining the integrity of the data set, we did not delete the samples of the data set, but used all of the samples of the imbalanced data set. Therefore, in addition to using accuracy, we also used some more reliable measures, such as F1, MCC, and AUC, and drew the ROC curve. We used these comprehensive indicators to better evaluate the performance of the model.
2.2. Characterization of Protein Evolution Information
We utilized the PSSM matrix to transform the protein evolution information in alphabetic form into a matrix in numerical form in the experiment. PSSM [
32] is able to translate protein sequences into numerical matrices and depict their biological evolutionary information [
33,
34,
35,
36,
37]. In the PSSM matrix, each protein can generate a
matrix
, which is mathematically described below:
here,
means the quantity of protein residues, 20 means the quantity of amino acid types, and the matrix element
denotes the probability of mutation of the ith residue to the
jth amino acid. In the experiment, we used position-specific iterated BLAST (PSI-BLAST) to generate the PSSM matrix of protein, and its download website is
http://blast.ncbi.nlm.nih.gov/blast.cgi (accessed date 1 May 2015). We set the parameter e-value and iterations of PSI-BLAST to the optimal 0.001 and 3, respectively, and searched for the protein sequences in the classical SwissProt database.
2.3. Protein Feature Extraction
In the experiment, Fast learning with Graph Convolutional Networks (FastGCN) is employed to extract the hidden features of the proteins [
38]. FastGCN is able to interpret graph nodes as independent identically distributed samples under a certain probability distribution and write the loss and each convolutional layer as an integral over the vertex embedding function, and to then evaluate the integral by defining a Monte Carlo approximation to the sample loss and sample gradient.
Suppose the probability space
correlates with the vertex set
of graph
. For a subgraph
of a graph
, its vertices are i.i.d. samples of
obtained by the probability measure
. It is mathematically represented as follows.
where
and
are independent random variables of
, and
is the embedding function from the
layer. Loss
is the expected value of
embedded in
, which is expressed as follows:
Thus, the i.i.d. sample
of
is available to approximately estimate the integral transformation in the
layer, which is described below:
where
is
. The loss
is translatable to the following:
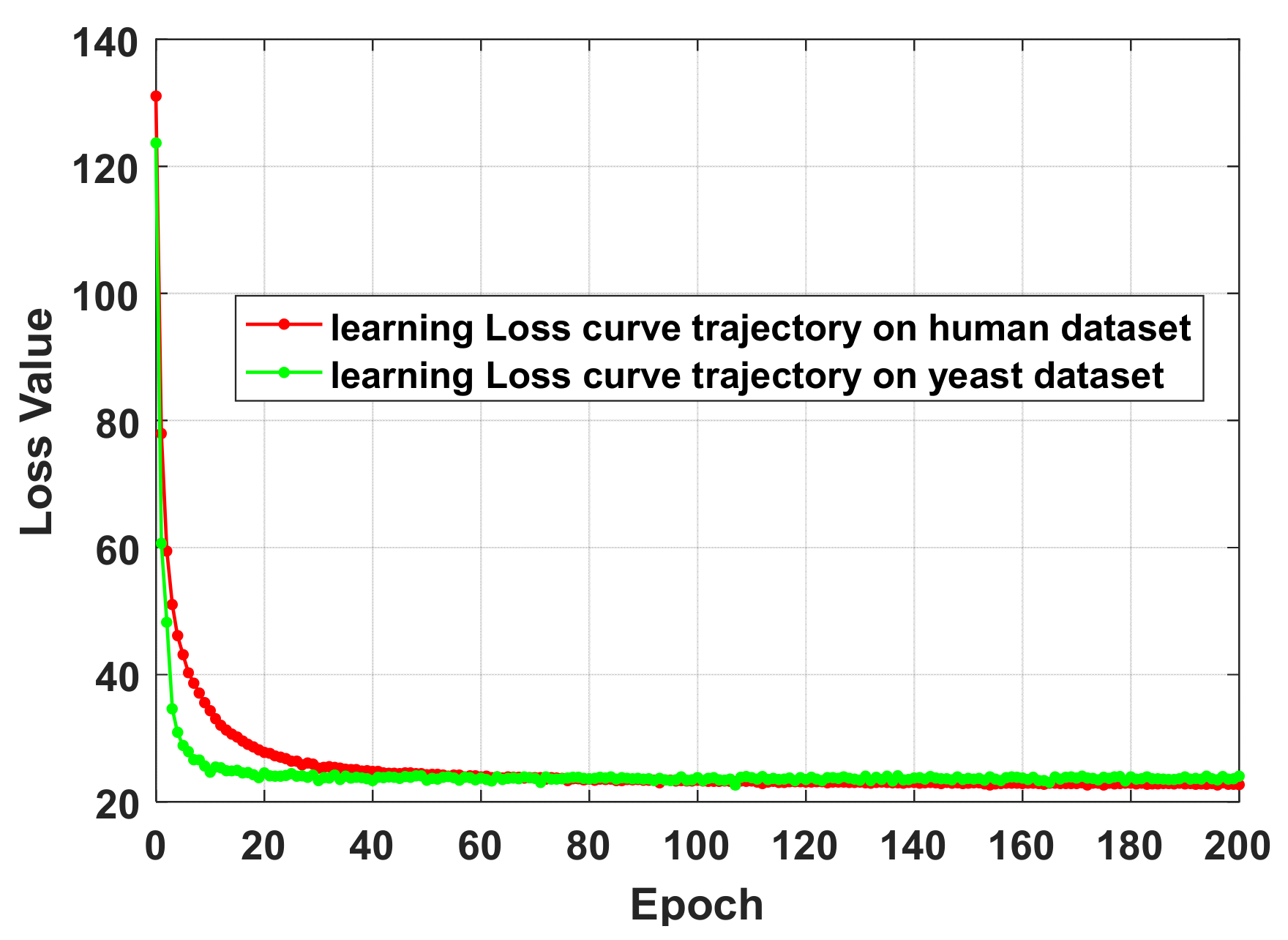
In the experiment, we verified the hyperparameter of FastGCN through the grid search method, and its optimization setting was as follows: the learning rate was 1e-1, the number of hidden layer neurons was 256, the number of iterations was 200, and the loss function was thr L2 regularization function. Specific experimental details can be found in
Supplementary Materials Table S1.
2.4. Interaction Prediction
We use a random forest (RF) classifier [
39,
40,
41] in the study to predict the interaction of the extracted feature data. RF contains multiple decision trees that classify new data by what they have learned in the data set using the following classification strategy.
- (a)
Construct sub-datasets by drawing samples from the dataset in a repeatable form according to the number of samples;
- (b)
Train decision trees based on these sub-datasets and obtain the results of each decision tree;
- (c)
Combine the results of all decision trees to obtain the final output using a minority–majority voting strategy.
4. Discussion
In this work, we designed an effective SIP prediction model SIPGCN based on protein amino acid sequences, combined with a deep learning GCN and RF classifier. We first used the PSSM matrix to obtain the evolutionary message of the amino acids, then extracted their hidden feature distributions using the GCN algorithm, and finally utilized the RF classifier on the gold standard data sets to determine whether there were interrelationships between them. SIPGCN shows an optimal performance after comparison with different models and previous methods. These excellent results indicate that SIPGCN has the ability to accurately predict SIP and can provide new insights for wet experiments.
There are two reasons SIPGCN performs so well. Firstly, SIPGCN makes full use of the evolutionary message of protein amino acids, which provides an excellent solution for the characterization of the sequence information; secondly, the feature extraction ability of deep learning GCN is quite impressive, which can extract the hidden feature distribution in protein network nodes as much as possible and can represent it as a numerical vector. With the support of these two advantages, SIPGCN naturally has a powerful prediction capability.
However, there are some limitations of SIPGCN. For example, SIPGCN only uses the sequence information of proteins and does not utilize their physicochemical information or 3D structure information, which needs to be further explored. Additionally, although the deep learning GCN method has a strong feature extraction capability, it has a high complexity and a large number of hyperparameters. How to better tune these hyperparameters to achieve optimal performance and reduce their complexity needs to be further resolved. These limitations motivate us to continuously improve the method and measure the performance of SIPGCN with higher requirements.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
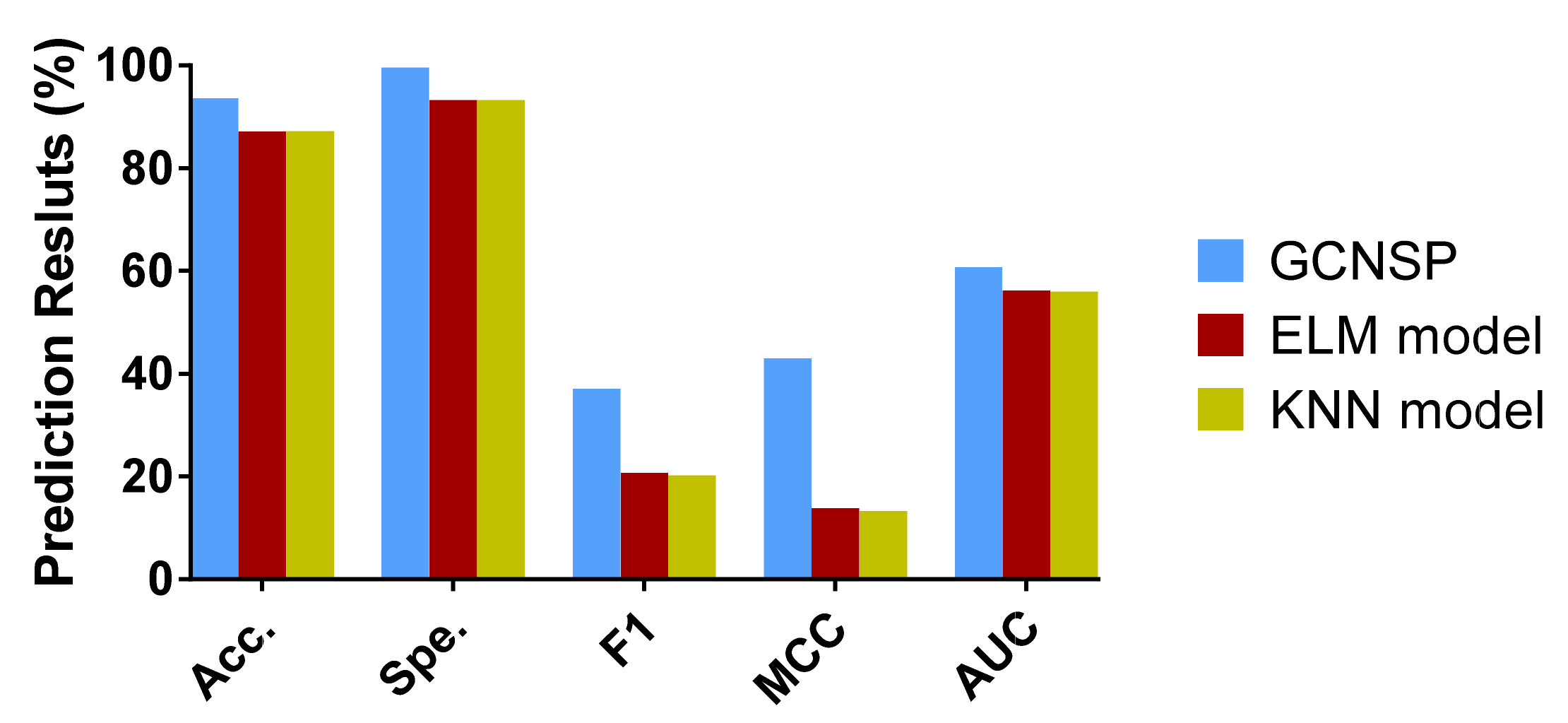{kind=link}
{kind=link}