

Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer

Abstract
1. Introduction
1.1. Development of CAR T Cells
1.2. Generations of CAR T Cells
1.3. Six FDA Approved CAR Cancer Immunotherapies
2. Multi-Specific CAR T Cells
2.1. Addressing the Challenge of Antigen Escape Using Car Technology
2.2. Development of TanCARs and LoopCARs
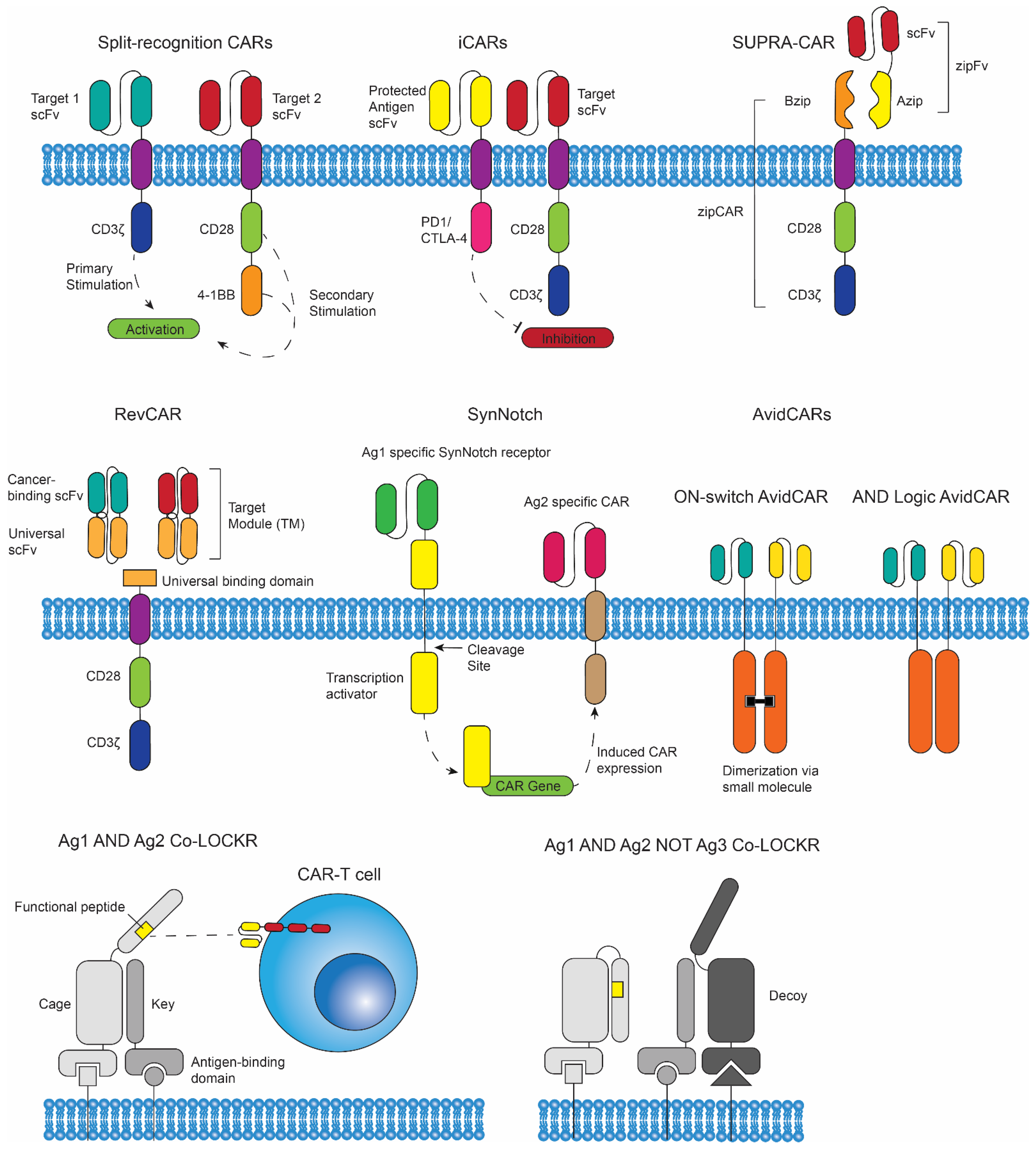
3. Logic Gated CAR T Cells
3.1. Introduction to Logic Gates
3.2. SUPRA CARs, RevCARs, SynNotch CAR T Cells, and AvidCARs
3.3. Co-LOCKRs
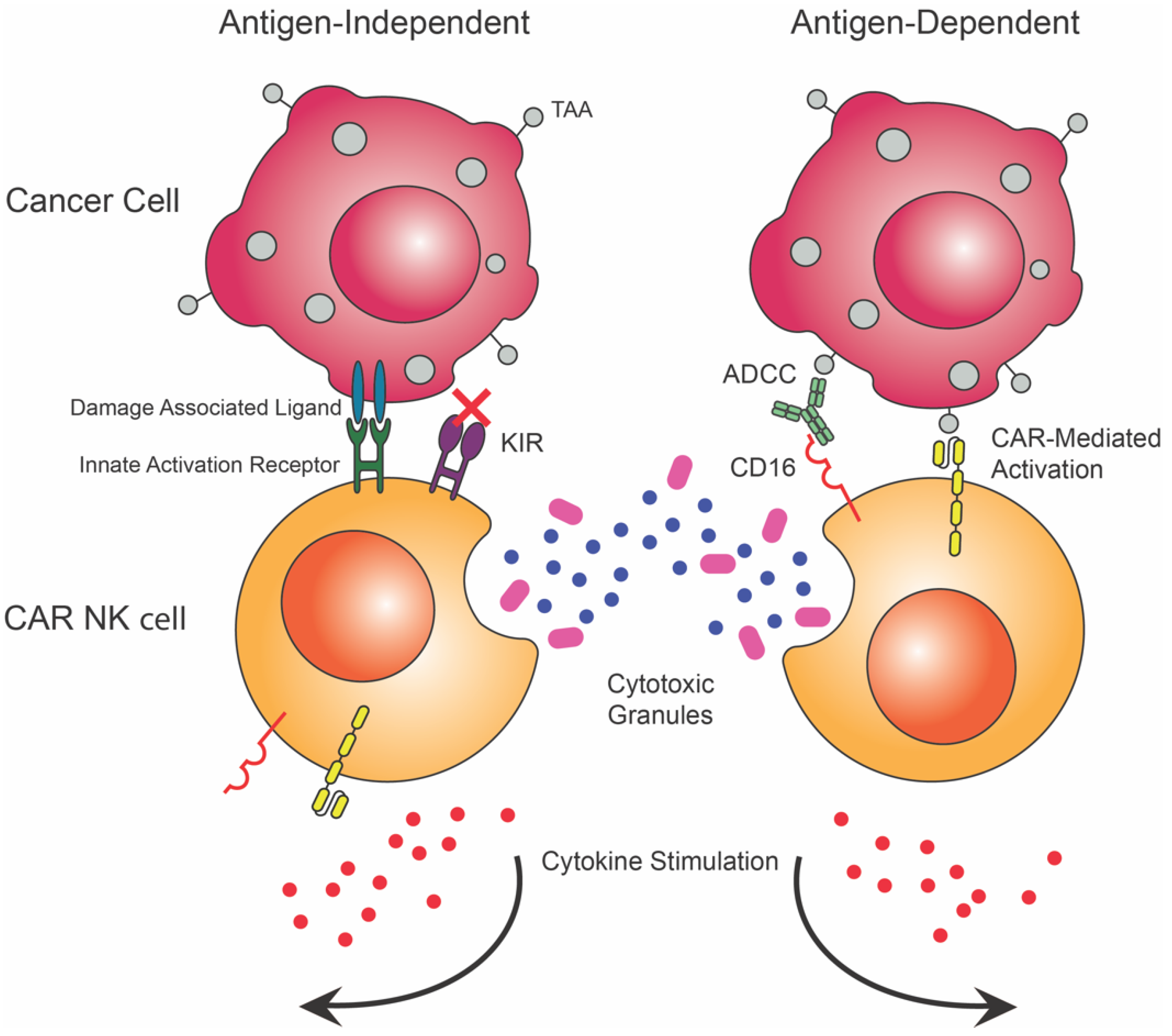
4. CAR NK Cells
4.1. Natural Killer Cells in Cancer Immunity
4.2. Availibility
4.3. Saftey
4.4. Efficacy
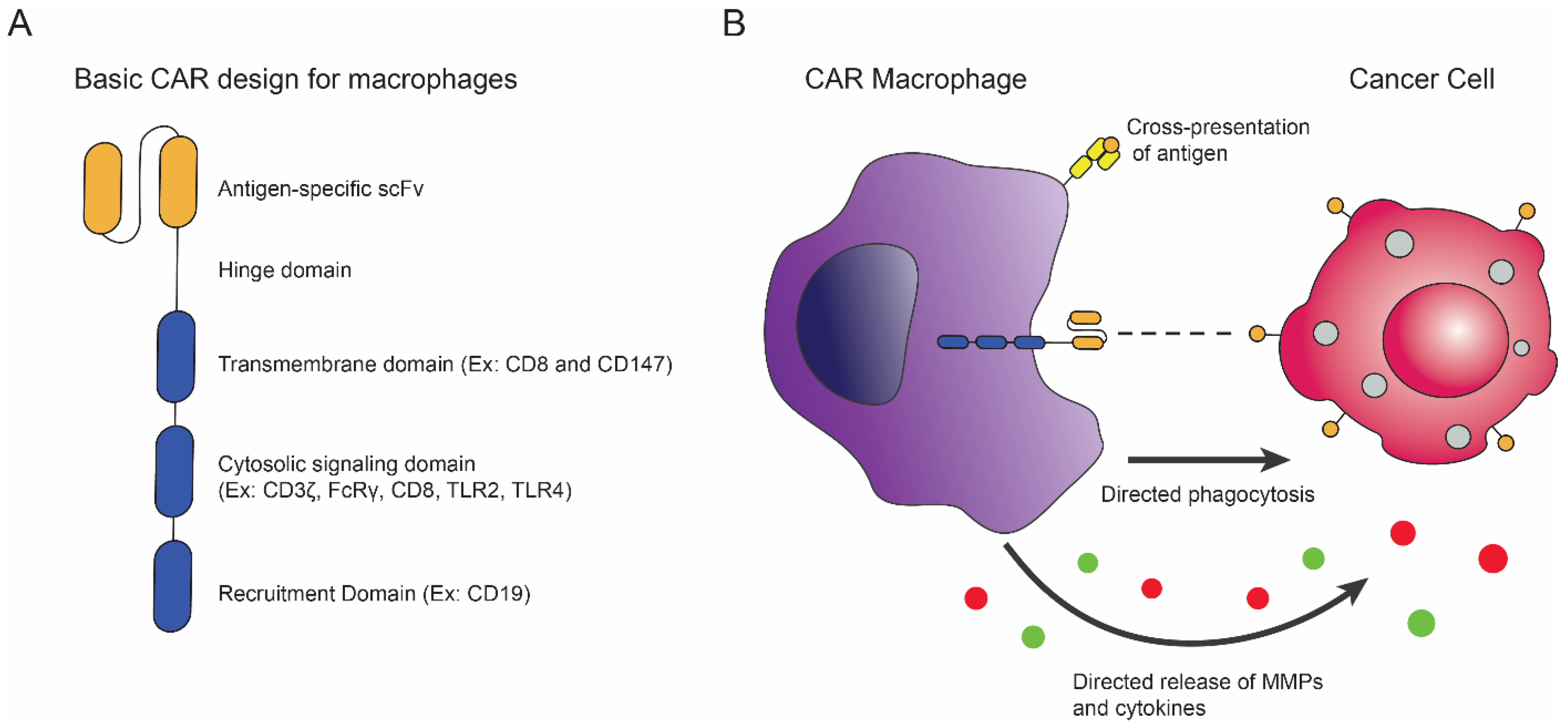
5. CAR Macrophages
5.1. Macrophages in Cancer Immunity
5.2. Introduction of CAR Macrophages and Recent Advances
5.3. Alternative CAR Macrophage Designs and Therapies
6. CARs beyond Cancer
6.1. CARs in Autoimmune Disease
6.2. Repurposing CAR T Cell Constructs for Autoimmune Disease
6.3. CAAR T Cells for Antibody Mediated Autoimmune Disease
6.4. CAR Treg Cells
6.5. CAR Use in Other Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 1–23. [Google Scholar] [CrossRef]
- Dapash, M.; Castro, B.; Hou, D.; Lee-Chang, C. Current immunotherapeutic strategies for the treatment of glioblastoma. Cancers 2021, 13, 4548. [Google Scholar] [CrossRef]
- Ottaviano, M.; de Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Arch. 2019, 474, 421–432. [Google Scholar] [CrossRef]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 2018, 175, 442–457.e23. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Boulch, M.; Cazaux, M.; Loe-Mie, Y.; Thibaut, R.; Corre, B.; Lemaître, F.; Grandjean, C.L.; Garcia, Z.; Bousso, P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci. Immunol. 2021, 6, eabd4344. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first CAR T therapy. Nat. Rev. Drug Discov. 2017, 16, 669. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Mohammadkhani, N.; Mahmoudi, R.; Gholizadeh, M.; Fakhr, E.; Cid-Arregui, A. TCR-like CARs and TCR-CARs targeting neoepitopes: An emerging potential. Cancer Gene Ther. 2021, 28, 581–589. [Google Scholar] [CrossRef]
- Mo, F.; Duan, S.; Jiang, X.; Yang, X.; Hou, X.; Shi, W.; Carlos, C.J.J.; Liu, A.; Yin, S.; Wang, W.; et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of CAR T cell therapy. molecular therapy. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.-X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed. Pharmacother. 2021, 146, 112512. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Moghanloo, E.; Mollanoori, H.; Talebi, M.; Pashangzadeh, S.; Faraji, F.; Hadjilooei, F.; Mahmoodzadeh, H. Remote controlling of CAR-T cells and toxicity management: Molecular switches and next generation CARs. Transl. Oncol. 2021, 14, 101070. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large b-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Ghosh, A.; Smith, M.; James, S.E.; Davila, M.L.; Velardi, E.; Argyropoulos, K.V.; Gunset, G.; Perna, F.; Kreines, F.M.; Levy, E.R.; et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med. 2017, 23, 242–249. [Google Scholar] [CrossRef]
- Vora, S.B.; Waghmare, A.; Englund, J.A.; Qu, P.; Gardner, R.A.; Hill, J.A. Infectious complications following CD19 chimeric antigen receptor t-cell therapy for children, adolescents, and young adults. Open Forum Infect. Dis. 2020, 7, ofaa121. [Google Scholar] [CrossRef]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 chimeric antigen receptor expression rescues t cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Porter, D. Cytokine release syndrome with chimeric antigen receptor t cell therapy. Biol. Blood Marrow Transplant. 2018, 25, e123–e127. [Google Scholar] [CrossRef] [PubMed]
- Freyer, C.W.; Porter, D.L. Cytokine release syndrome and neurotoxicity following CAR T-cell therapy for hematologic malignancies. J. Allergy Clin. Immunol. 2020, 146, 940–948. [Google Scholar] [CrossRef]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current progress in CAR-T cell therapy for solid tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef]
- Abramson, J.S. Anti-CD19 CAR t-cell therapy for b-cell non-hodgkin lymphoma. Transfus. Med. Rev. 2019, 34, 29–33. [Google Scholar] [CrossRef]
- Weisel, K.; Martin, T.; Krishnan, A.; Jagannath, S.; Londhe, A.; Nair, S.; Diels, J.; Vogel, M.; Schecter, J.M.; Banerjee, A.; et al. Comparative efficacy of ciltacabtagene autoleucel in CARTITUDE-1 vs physician’s choice of therapy in the long-term follow-up of POLLUX, CASTOR, and EQUULEUS clinical trials for the treatment of patients with relapsed or refractory multiple myeloma. Clin. Drug Investig. 2022, 42, 29–41. [Google Scholar] [CrossRef]
- Nadler, L.M.; Anderson, K.C.; Marti, G.; Bates, M.; Park, E.; Daley, J.F.; Schlossman, S.F. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J. Immunol. 1983, 131, 244–250. [Google Scholar]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells—Behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first BCMA-targeted CAR-T cell therapy. Nat. Rev. Drug Discov. 2021, 20, 332. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef]
- Walsh, Z.; Ross, S.; Fry, T.J. Multi-specific CAR targeting to prevent antigen escape. Curr. Hematol. Malign- Rep. 2019, 14, 451–459. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Cronk, R.J.; Zurko, J.; Shah, N.N. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers 2020, 12, 2523. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Shalabi, H.; Qin, H.; Su, A.; Yates, B.; Wolters, P.L.; Steinberg, S.M.; Ligon, J.A.; Silbert, S.; DéDé, K.; Benzaoui, M.; et al. CD19/22 CAR T-cells in children and young adults with B-ALL: Phase I results and development of a novel bicistronic CAR. Blood 2022, in press. [CrossRef] [PubMed]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef]
- van der Schans, J.J.; van de Donk, N.; Mutis, T. Dual targeting to overcome current challenges in multiple myeloma CAR T-cell treatment. Front. Oncol. 2020, 10, 1362. [Google Scholar] [CrossRef]
- Savanur, M.A.; Weinstein-Marom, H.; Gross, G. Implementing logic gates for safer immunotherapy of cancer. Front. Immunol. 2021, 12, 780399. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–Based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Okuma, A.; Sofjan, K.; Lee, S.; Collins, J.J.; Wong, W.W. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 2021, 12, 792. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Loureiro, L.R.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. OncoImmunology 2020, 9, 1785608. [Google Scholar] [CrossRef]
- Kittel-Boselli, E.; Soto, K.E.G.; Loureiro, L.R.; Hoffmann, A.; Bergmann, R.; Arndt, C.; Koristka, S.; Mitwasi, N.; Kegler, A.; Bartsch, T.; et al. Targeting acute myeloid leukemia using the RevCAR platform: A programmable, switchable and combinatorial strategy. Cancers 2021, 13, 4785. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Boyken, S.E.; Salter, A.I.; Bruffey, J.; Rajan, A.; Langan, R.A.; Olshefsky, A.; Muhunthan, V.; Bick, M.J.; Gewe, M.; et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 2020, 369, 1637–1643. [Google Scholar] [CrossRef]
- Vaněk, O.; Kalousková, B.; Abreu, C.; Nejadebrahim, S.; Skořepa, O. Natural killer cell-based strategies for immunotherapy of cancer. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 129, pp. 91–133. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Daher, R. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Yang, Y.; Li, W.; Tan, Y.; Guo, W.; Ao, L.; He, X.; Wu, X.; Xia, J.; Xu, X.; et al. Anti-αFR CAR-engineered NK-92 cells display potent cytotoxicity against αFR-positive ovarian cancer. J. Immunother. 2019, 42, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; de Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK cells: A new non-viral method allowing high efficient transfection and strong tumor cell killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front. Immunol. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front. Immunol. 2020, 10, 3123. [Google Scholar] [CrossRef]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Upshaw, J.L.; Arneson, L.N.; A Schoon, R.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef]
- Ebrahimiyan, H.; Tamimi, A.; Shokoohian, B.; Minaei, N.; Memarnejadian, A.; Hossein-Khannazer, N.; Hassan, M.; Vosough, M. Novel insights in CAR-NK cells beyond CAR-T cell technology; Promising advantages. Int. Immunopharmacol. 2022, 106, 108587. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Cienfuegos-Jimenez, O.; Vazquez-Garza, E.; Rojas-Martinez, A. CAR-NK cells for cancer therapy: Molecular redesign of the innate antineoplastic response. Curr. Gene Ther. 2021, 22, 303–318. [Google Scholar] [CrossRef]
- Sheng, L.; Mu, Q.; Wu, X.; Yang, S.; Zhu, H.; Wang, J.; Lai, Y.; Wu, H.; Sun, Y.; Hu, Y.; et al. Cytotoxicity of donor natural killer cells to allo-reactive t cells are related with acute graft-vs.-host-disease following allogeneic stem cell transplantation. Front. Immunol. 2020, 11, 1534. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Matosevic, S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Cooper, M.A. Harnessing NK cell memory for cancer immunotherapy. Trends Immunol. 2016, 37, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Paust, S.; von Andrian, U.H. Natural killer cell memory. Nat. Immunol. 2011, 12, 500–508. [Google Scholar] [CrossRef]
- Arachchige, A.S.P.M. Human NK cells: From development to effector functions. Innate Immun. 2021, 27, 212–229. [Google Scholar] [CrossRef]
- Zuo, W.; Yu, X.X.; Liu, X.F.; Chang, Y.J.; Wang, Y.; Zhang, X.H.; Xu, L.P.; Liu, K.Y.; Zhao, X.S.; Huang, X.J.; et al. The interaction of HLA-C1/KIR2DL2/L3 promoted KIR2DL2/L3 single-positive/NKG2C-positive natural killer cell reconstitution, raising the incidence of aGVHD after hematopoietic stem cell transplantation. Front Immunol. 2022, 13, 814334. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current perspectives on “off-the-shelf” allogeneic NK and CAR-NK cell therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.; Yu, J.; Varshney, M.; Inzunza, J.; Nalvarte, I. Hematopoietic Stem Cell- and induced pluripotent stem cell-derived CAR-NK cells as reliable cell-based therapy solutions. Stem Cells Transl. Med. 2021, 10, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef]
- Caruso, H.G.; Tanaka, R.; Liang, J.; Ling, X.; Sabbagh, A.; Henry, V.K.; Collier, T.L.; Heimberger, A.B. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J. Neurooncol. 2019, 145, 429–439. [Google Scholar] [CrossRef]
- Klöß, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of human NK cell manufacturing: Fully automated separation, improved Ex Vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Tanaka, J.; Miller, J.S. Recent progress in and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. 2020, 44, 100678. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front. Immunol. 2017, 8, 465. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef]
- Lee, M.Y.; Robbins, Y.; Sievers, C.; Friedman, J.; Sater, H.A.; E Clavijo, P.; Judd, N.; Tsong, E.; Silvin, C.; Soon-Shiong, P.; et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J. Immunother. Cancer 2021, 9, e002128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK cells for cancer immunotherapy: From bench to bedside. Biomark. Res. 2022, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J. Immunol. Res. 2021, 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, X.; Stojanovic, A.; Zhang, Q.; Wincher, M.; Bühler, L.; Arnold, A.; Correia, M.P.; Winkler, M.; Koch, P.-S.; et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α unleashes NK cell activity. Immunity 2020, 52, 1075–1087.e8. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell. Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Jaguin, M.; Houlbert, N.; Fardel, O.; Lecureur, V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell. Immunol. 2013, 281, 51–61. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lawrence, T.; A Hamilton, J.; Cook, A. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: Implications for CSF blockade in inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Kastelein, R.; Menon, S.; Andrade, S.; Coffman, R.L. Modulation of murine macrophage function by IL-13. J. Immunol. 1993, 151, 7151–7160. [Google Scholar] [PubMed]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Toi, M.; Saji, H.; Muta, M.; Bando, H.; Kuroi, K.; Koike, M.; Inadera, H.; Matsushima, K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000, 6, 3282–3289. [Google Scholar] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef]
- Kim, J.; Modlin, R.; Moy, R.L.; Dubinett, S.M.; McHugh, T.; Nickoloff, B.J.; Uyemura, K. IL-10 production in cutaneous basal and squamous cell carcinomas. A mechanism for evading the local T cell immune response. J. Immunol. 1995, 155, 2240–2247. [Google Scholar]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; van Damme, J.; Mantovani, A. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef]
- Loercher, A.E.; Nash, M.A.; Kavanagh, J.J.; Platsoucas, C.D.; Freedman, R.S. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J. Immunol. 1999, 163, 6251–6260. [Google Scholar]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 2019, 10, 1611. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lim, S.Y.; Gordon-Weeks, A.N.; Tapmeier, T.T.; Im, J.H.; Cao, Y.; Beech, J.; Allen, D.; Smart, S.; Muschel, R.J. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 2013, 57, 829–839. [Google Scholar] [CrossRef]
- Peña, C.G.; Nakada, Y.; Saatcioglu, H.D.; Aloisio, G.M.; Cuevas, I.; Zhang, S.; Miller, D.S.; Lea, J.S.; Wong, K.-K.; de Berardinis, R.J.; et al. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J. Clin. Investig. 2015, 125, 4063–4076. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Ewell, Z.D.; Bennion, K.A.; Wang, G.; Skidmore, J.; Lum, D.; Boyer, M.; O’Neill, K.L. MOTO-CARs (TM): A novel macrophage-based chimeric antigen receptor technology. Cancer Res. 2020, 80 (Suppl. 16), 3254. [Google Scholar] [CrossRef]
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Front. Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef]
- Bartok, E.; Hartmann, G. Immune sensing mechanisms that discriminate self from altered self and foreign nucleic acids. Immunity 2020, 53, 54–77. [Google Scholar] [CrossRef]
- Bobadilla, S.; Sunseri, N.; Landau, N.R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2012, 20, 514–520. [Google Scholar] [CrossRef]
- Ohtani, Y.; Ross, K.; Dandekar, A.; Gabbasov, R.; Klichinsky, M. 128 Development of an M1-polarized, non-viral chimeric antigen receptor macrophage (CAR-M) platform for cancer immunotherapy. J. Immunother. Cancer 2020, 8, A141. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Wang, N.; Liu, J.; Cai, Y.; Ren, M.; Li, Z. A simple and efficient method for the generation of a porcine alveolar macrophage cell line for high-efficiency Porcine reproductive and respiratory syndrome virus 2 infection. J. Virol. Methods 2019, 274, 113727. [Google Scholar] [CrossRef] [PubMed]
- Moradian, H.; Roch, T.; Lendlein, A.; Gossen, M. mRNA transfection-induced activation of primary human monocytes and macrophages: Dependence on carrier system and nucleotide modification. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Hickey, M. Mechanisms of lymphocyte migration in autoimmune disease. Tissue Antigens 2005, 66, 163–172. [Google Scholar] [CrossRef]
- Mazzi, M.T.; Hajdu, K.L.; Ribeiro, P.R.; Bonamino, M.H. CAR-T cells leave the comfort zone: Current and future applications beyond cancer. Immunother. Adv. 2021, 1, ltaa006. [Google Scholar] [CrossRef]
- Sang, A.; Zheng, Y.-Y.; Morel, L. Contributions of B cells to lupus pathogenesis. Mol. Immunol. 2014, 62, 329–338. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.; Hsieh, H.-J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2009, 62, 222–233. [Google Scholar] [CrossRef]
- Mendez, L.M.G.; Cascino, M.D.; Garg, J.; Katsumoto, T.R.; Brakeman, P.; Dall’Era, M.; Looney, R.J.; Rovin, B.; Dragone, L.; Brunetta, P. Peripheral blood B cell depletion after rituximab and complete response in lupus nephritis. Clin. J. Am. Soc. Nephrol. 2018, 13, 1502–1509. [Google Scholar] [CrossRef]
- Kotagiri, P.; Martin, A.; Hughes, P.; Becker, G.; Nicholls, K. Single-dose rituximab in refractory lupus nephritis. Intern. Med. J. 2016, 46, 899–901. [Google Scholar] [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.-U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Arhontoulis, D.; Wertheim, G.; Capobianchi, J.; Callahan, C.A.; Ellebrecht, C.T.; Obstfeld, A.E.; Lacey, S.F.; Melenhorst, J.J.; Nazimuddin, F.; et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 2016, 128, 360–370. [Google Scholar] [CrossRef]
- Fishman, S.; Lewis, M.D.; Siew, L.K.; De Leenheer, E.; Kakabadse, D.; Davies, J.; Ziv, D.; Margalit, A.; Karin, N.; Gross, G.; et al. Adoptive transfer of mRNA-transfected T cells redirected against diabetogenic CD8 T cells can prevent diabetes. Mol. Ther. 2017, 25, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.S.; Fishman, S.; Margalit, A.; Siew, L.K.; Chapman, S.; Wen, L.; Gross, G.; Wong, F.S. Developing a novel model system to target insulin-reactive CD8 T cells. Ann. N. Y. Acad. Sci. 2008, 1150, 54–58. [Google Scholar] [CrossRef]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2018, 96, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Crawford, F.; Yu, L.; Michels, A.; Nakayama, M.; Davidson, H.W.; Kappler, J.W.; Eisenbarth, G.S. Monoclonal antibody blocking the recognition of an insulin peptide–MHC complex modulates type 1 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 2656–2661. [Google Scholar] [CrossRef]
- Treanor, B. B-cell receptor: From resting state to activate. Immunology 2012, 136, 21–27. [Google Scholar] [CrossRef]
- Hammers, C.M.; Stanley, J.R. Mechanisms of disease: Pemphigus and bullous pemphigoid. Annu. Rev. Pathol. 2016, 11, 175–197. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef]
- Lee, J.; Lundgren, D.K.; Mao, X.; Manfredo-Vieira, S.; Nunez-Cruz, S.; Williams, E.F.; Assenmacher, C.-A.; Radaelli, E.; Oh, S.; Wang, B.; et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J. Clin. Investig. 2020, 130, 6317–6324. [Google Scholar] [CrossRef]
- Oh, S.; O’Connor, K.; Payne, A.Y. MuSK Chimeric Autoantibody receptor (CAAR) T cells for antigen specific immunotherapy of Myasthenia Gravis (2769). Neurology 2020, 94, 2769. [Google Scholar]
- Scott, D.W.; Pratt, K.P.; Miao, C.H. Progress toward inducing immunologic tolerance to factor VIII. Blood 2013, 121, 4449–4456. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; Berg, H.M.V.D.; Oldenburg, J.; Astermark, J.; de Groot, P.G.; Margaglione, M.; Thompson, A.R.; van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, K.; Scott, D.W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018, 2, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Sadeqi Nezhad, M.; Seifalian, A.; Bagheri, N.; Yaghoubi, S.; Karimi, M.H.; Adbollahpour-Alitappeh, M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: How close are we to the treatment? Front Immunol. 2020, 11, 603237. [Google Scholar] [CrossRef]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef]
- Beheshti, S.A.; Shamsasenjan, K.; Ahmadi, M.; Abbasi, B. CAR Treg: A new approach in the treatment of autoimmune diseases. Int. Immunopharmacol. 2022, 102, 108409. [Google Scholar] [CrossRef]
- Beres, A.; Komorowski, R.; Mihara, M.; Drobyski, W.R. Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin. Cancer Res. 2011, 17, 3969–3983. [Google Scholar] [CrossRef]
- Bhela, S.; Varanasi, S.K.; Jaggi, U.; Sloan, S.S.; Rajasagi, N.K.; Rouse, B.T. The plasticity and stability of regulatory T cells during viral-induced inflammatory lesions. J. Immunol. 2017, 199, 1342–1352. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Elinav, E.; Adam, N.; Waks, T.; Eshhar, Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 2009, 136, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S.I. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflamm. 2012, 9, 112. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Yoon, J.; Schmidt, A.; Zhang, A.-H.; Königs, C.; Kim, Y.C.; Scott, D.W. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood 2017, 129, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Pohl, A.D.P.; Venkatesha, S.H.; Zhang, A.-H.; Scott, D.W. Suppression of FVIII-specific memory B cells by chimeric BAR receptor-engineered natural regulatory T cells. Front. Immunol. 2020, 11, 693. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef]
- Roberts, M.R.; Qin, L.; Zhang, D.; Smith, D.H.; Tran, A.C.; Dull, T.J.; Groopman, J.E.; Capon, D.J.; Byrn, R.A.; Finer, M.H. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood 1994, 84, 2878–2889. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.; et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Kuhlmann, A.-S.; Peterson, C.W.; Kiem, H.-P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS 2018, 13, 446–453. [Google Scholar] [CrossRef]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-based bispecific chimeric antigen receptor designed for enhanced anti-HIV potency and absence of HIV entry receptor activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef] [PubMed]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T cell therapy beyond oncology: Autoimmune diseases and viral infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Badeti, S.; Geng, K.; Liu, D. Efficacy of targeting SARS-CoV-2 by CAR-NK Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kürschner, T.; Schulze, A.; Urban, S.; Krönke, M.; Abken, H.; Protzer, U. T Cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.L.; Shum, T.; Tashiro, H.; Barzi, M.; Yi, Z.; Whitten-Bauer, C.; Legras, X.; Bissig-Choisat, B.; Garaigorta, U.; Gottschalk, S.; et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 2018, 20, 697–705. [Google Scholar] [CrossRef]
- Sautto, G.A.; Wisskirchen, K.; Clementi, N.; Castelli, M.; Diotti, R.A.; Graf, J.; Clementi, M.; Burioni, R.; Protzer, U.; Mancini, N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 2016, 65, 512–523. [Google Scholar] [CrossRef]
- Proff, J.; Walterskirchen, C.; Brey, C.; Geyeregger, R.; Full, F.; Ensser, A.; Lehner, M.; Holter, W. Cytomegalovirus-infected cells resist T cell mediated killing in an HLA-recognition independent manner. Front. Microbiol. 2016, 7, 844. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475. [Google Scholar] [CrossRef]
- Tang, X.; Tang, Q.; Mao, Y.; Huang, X.; Jia, L.; Zhu, J.; Feng, Z. CD137 Co-stimulation improves the antitumor effect of LMP1-specific chimeric antigen receptor T cells in vitro and in vivo. OncoTargets Ther. 2019, 12, 9341–9350. [Google Scholar] [CrossRef]
- Talbot, S.J.; Blair, N.F.; McGill, N.; Ligertwood, Y.; Dutia, B.M.; Johannessen, I. An influenza virus M2 protein specific chimeric antigen receptor modulates influenza A/WSN/33 H1N1 infection in vivo. Open Virol. J. 2013, 7, 28–36. [Google Scholar] [CrossRef][Green Version]
- Kumaresan, P.R.; Manuri, P.R.; Albert, N.D.; Maiti, S.; Singh, H.; Mi, T.; Roszik, J.; Rabinovich, B.; Olivares, S.; Krishnamurthy, J.; et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl. Acad. Sci. USA 2014, 111, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.J.; Johansen, A.K.Z.; Molkentin, J.D. CARdiac immunotherapy: T cells engineered to treat the fibrotic heart. Mol. Ther. 2019, 27, 1869–1871. [Google Scholar] [CrossRef]
- Baraldo, S.; Oliani, K.L.; Turato, G.; Zuin, R.; Saetta, M. The role of lymphocytes in the pathogenesis of asthma and COPD. Curr. Med. Chem. 2007, 14, 2250–2256. [Google Scholar] [CrossRef]
- Ward, D.E.; Fay, B.L.; Adejuwon, A.; Han, H.; Ma, Z. Chimeric antigen receptors based on low affinity mutants of FcεRI re-direct T cell specificity to cells expressing membrane IgE. Front. Immunol. 2018, 9, 2231. [Google Scholar] [CrossRef]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric antigen receptor-redirected regulatory T cells suppress experimental allergic airway inflammation, a model of asthma. Front. Immunol. 2017, 8, 1125. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAR Construct | Mechanism | Benefits | Drawbacks | FDA-approvedor Highest Clinical trial |
| CAR T cell |
|
|
| Six FDA-approved treatments |
| Multi-specific CAR T cell |
|
|
| Phase II |
| CAR NK cell |
|
|
| Phase II |
| CAR macrophage |
|
|
| Phase I |
| CAAR T cell |
|
|
| Phase I |
| CAR Treg cell |
|
|
| Preclinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, C.; Haynie, C.; Cheever, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. https://doi.org/10.3390/biomedicines10071493
Moreno C, Haynie C, Cheever A, Weber KS. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines. 2022; 10(7):1493. https://doi.org/10.3390/biomedicines10071493
Chicago/Turabian StyleMoreno, Carlos, Christopher Haynie, Abigail Cheever, and K. Scott Weber. 2022. "Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer" Biomedicines 10, no. 7: 1493. https://doi.org/10.3390/biomedicines10071493
APA StyleMoreno, C., Haynie, C., Cheever, A., & Weber, K. S. (2022). Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines, 10(7), 1493. https://doi.org/10.3390/biomedicines10071493