
Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases


Abstract
1. Introduction
2. Signature Cytokines and Associated Molecular Pathways Involved in the Idiopathic Counterparts of PRs Caused by Biologic Agents
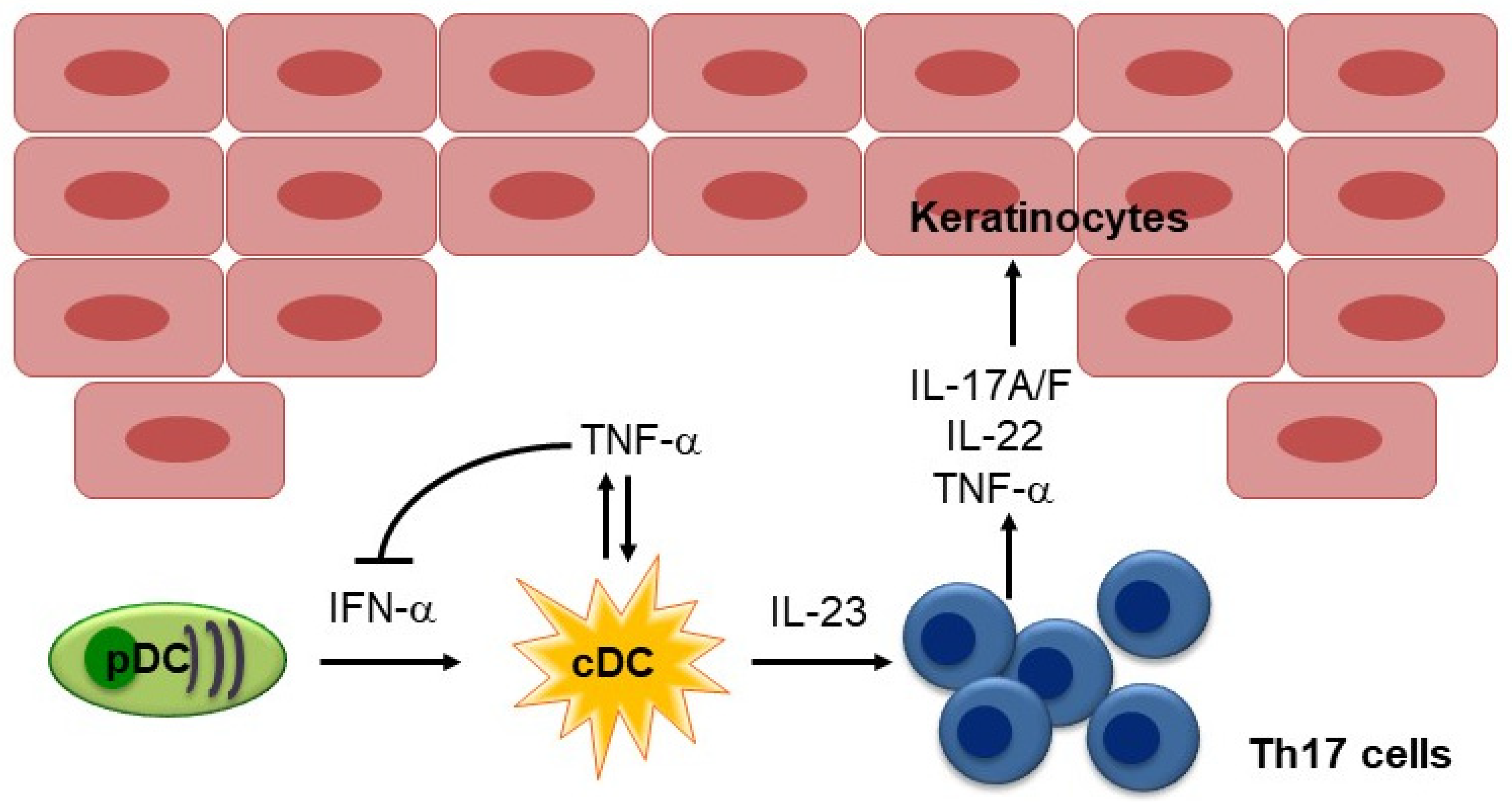
2.1. IL-17 and IL-23 in Psoriasis
2.2. IL-4, IL-13, and IL-5 in Atopic Dermatitis
2.3. Type I IFNs in Systemic Lupus Erythematosus
3. Paradoxical Reactions Caused by Each Type of Biologic Agent
3.1. TNF-α Inhibitors
3.1.1. Psoriasiform Reactions
Mechanism Responsible for Psoriasiform Reactions Induced by TNF-α Inhibitors
3.1.2. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by TNF-α Inhibitors
3.1.3. Lupus-Like Reactions
Clinical Features of Lupus-like Reactions Induced by TNF-α Inhibitors
Mechanism Responsible for Lupus-like Reactions Induced by TNF-α Inhibitors
3.2. IL-12/23 p40 Inhibitors
3.2.1. Psoriasiform Reactions
3.2.2. Eczematous Reactions
3.2.3. Ustekinumab for Skin Reactions Associated with TNF-α Inhibitors
3.3. IL-17 Inhibitors
3.3.1. Psoriasiform Reactions
3.3.2. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by IL-17 Inhibitors
3.4. IL-23p19 Inhibitors
3.4.1. Eczematous Reactions
Mechanism Responsible for Eczematous Reactions Induced by IL-23p19 Inhibitors
3.5. IL-4Rα Inhibitors
3.5.1. Psoriasiform Reactions
Mechanism Responsible for Psoriasiform Reactions Induced by IL-4Rα Inhibitors
3.5.2. Eczematous Reactions
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murphy, M.J.; Cohen, J.M.; Vesely, M.D.; Damsky, W. Paradoxical eruptions to targeted therapies in dermatology: A systematic review and analysis. J. Am. Acad. Dermatol. 2020, 86, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; De Simone, C.; Genovese, G.; Berti, E.; Cugno, M.; Marzano, A.V. Paradoxical Skin Reactions to Biologics in Patients with Rheumatologic Disorders. Front. Pharmacol. 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Pérez-De-Lis, M.; Retamozo, S.; Flores-Chávez, A.; Kostov, B.; Perez-Alvarez, R.; Brito-Zerón, P.; Ramos-Casals, M. Autoimmune diseases induced by biological agents. A review of 12,731 cases (BIOGEAS Registry). Expert Opin. Drug Saf. 2017, 16, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, K.; Eyerich, S. Immune response patterns in non-communicable inflammatory skin diseases. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; McInnes, I.B.; Neurath, M.F. Reframing Immune-Mediated Inflammatory Diseases through Signature Cytokine Hubs. N. Engl. J. Med. 2021, 385, 628–639. [Google Scholar] [CrossRef]
- Braegelmann, C.; Niebel, D.; Wenzel, J. Targeted Therapies in Autoimmune Skin Diseases. J. Investig. Dermatol. 2022, 142, 969–975.e7. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Wenzel, J.; Tuting, T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. J. Investig. Dermatol. 2008, 128, 2392–2402. [Google Scholar] [CrossRef]
- Eyerich, S.; Onken, A.T.; Weidinger, S.; Franke, A.; Nasorri, F.; Pennino, D.; Grosber, M.; Pfab, F.; Schmidt-Weber, C.B.; Mempel, M.; et al. Mutual Antagonism of T Cells Causing Psoriasis and Atopic Eczema. N. Engl. J. Med. 2011, 365, 231–238. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis Vulgaris Lesions Contain Discrete Populations of Th1 and Th17 T Cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef]
- Croxford, A.L.; Karbach, S.; Kurschus, F.C.; Wörtge, S.; Nikolaev, A.; Yogev, N.; Klebow, S.; Schüler, R.; Reissig, S.; Piotrowski, C.; et al. IL-6 Regulates Neutrophil Microabscess Formation in IL-17A-Driven Psoriasiform Lesions. J. Investig. Dermatol. 2014, 134, 728–735. [Google Scholar] [CrossRef]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Piskin, G.; Sylva-Steenland, R.M.R.; Bos, J.D.; Teunissen, M.B.M. In Vitro and In Situ Expression of IL-23 by Keratinocytes in Healthy Skin and Psoriasis Lesions: Enhanced Expression in Psoriatic Skin. J. Immunol. 2006, 176, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deshpande, M.; Grisotto, M.; Smaldini, P.; Garcia, R.; He, Z.; Gulko, P.S.; Lira, S.A.; Furtado, G.C. Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci. Rep. 2020, 10, 8259. [Google Scholar] [CrossRef]
- Lowes, M.A.; Chamian, F.; Abello, M.V.; Fuentes-Duculan, J.; Lin, S.-L.; Nussbaum, R.; Novitskaya, I.; Carbonaro, H.; Cardinale, I.; Kikuchi, T.; et al. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc. Natl. Acad. Sci. USA 2005, 102, 19057–19062. [Google Scholar] [CrossRef]
- Boyman, O.; Hefti, H.P.; Conrad, C.; Nickoloff, B.J.; Suter, M.; Nestle, F.O. Spontaneous Development of Psoriasis in a New Animal Model Shows an Essential Role for Resident T Cells and Tumor Necrosis Factor-α. J. Exp. Med. 2004, 199, 731–736. [Google Scholar] [CrossRef]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.-J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Hamid, Q.; Boguniewicz, M.; Leung, D.Y. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J. Clin. Investig. 1994, 94, 870–876. [Google Scholar] [CrossRef]
- Lee, G.R.; Flavell, R.A. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int. Immunol. 2004, 16, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.S.; Robinson, N.; Xu, L. Expression of Interleukin-4 in the Epidermis of Transgenic Mice Results in a Pruritic Inflammatory Skin Disease: An Experimental Animal Model to Study Atopic Dermatitis. J. Investig. Dermatol. 2001, 117, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Oh, M.H.; Oh, S.Y.; Schroeder, J.T.; Glick, A.B.; Zhu, Z. Transgenic Expression of Interleukin-13 in the Skin Induces a Pruritic Dermatitis and Skin Remodeling. J. Investig. Dermatol. 2009, 129, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; Strong, C.D.G.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune Interferon in the Circulation of Patients with Autoimmune Disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Peterson, M.G.E.; Crow, M.K. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005, 52, 1491–1503. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Rönnblom, L.E.; Alm, G.V.; Öberg, K.E. Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 1990, 227, 207–210. [Google Scholar] [CrossRef]
- Niewold, T.B.; Swedler, W.I. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol. 2005, 24, 178–181. [Google Scholar] [CrossRef]
- Baccala, R.; Gonzalez-Quintial, R.; Blasius, A.L.; Rimann, I.; Ozato, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc. Natl. Acad. Sci. USA 2013, 110, 2940–2945. [Google Scholar] [CrossRef] [PubMed]
- Lövgren, T.; Eloranta, M.-L.; Båve, U.; Alm, G.V.; Rönnblom, L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Raber, M.-L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, F.; Tagaya, Y.; Ozato, K.; Asada, H. Essential Requirement for IFN Regulatory Factor 7 in Autoantibody Production but Not Development of Nephritis in Murine Lupus. J. Immunol. 2016, 197, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, F.; Tagaya, Y.; Ozato, K.; Horie, K.; Asada, H. Inflammatory monocyte-derived dendritic cells mediate autoimmunity in murine model of systemic lupus erythematosus. J. Transl. Autoimmun. 2020, 3, 100060. [Google Scholar] [CrossRef]
- Pasadyn, S.R.; Knabel, D.; Fernandez, A.P.; Warren, C.B. Cutaneous adverse effects of biologic medications. Cleve. Clin. J. Med. 2020, 87, 288–299. [Google Scholar] [CrossRef]
- Moustou, A.-E.; Matekovits, A.; Dessinioti, C.; Antoniou, C.; Sfikakis, P.P.; Stratigos, A.J. Cutaneous side effects of anti–tumor necrosis factor biologic therapy: A clinical review. J. Am. Acad. Dermatol. 2009, 61, 486–504. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Zaba, L.C.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030.e395. [Google Scholar] [CrossRef]
- Baeten, D.; Kruithof, E.; Van den Bosch, F.; Van den Bossche, N.; Herssens, A.; Mielants, H.; De Keyser, F.; Veys, E.M. Systematic safety follow up in a cohort of 107 patients with spondyloarthropathy treated with infliximab: A new perspective on the role of host defence in the pathogenesis of the disease? Ann. Rheum. Dis. 2003, 62, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Verea, M.M.; Del Pozo, J.; Yebra-Pimentel, M.T.; Porta, A.; Fonseca, E. Psoriasiform Eruption Induced by Infliximab. Ann. Pharmacother. 2004, 38, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Thurber, M.; Feasel, A.; Stroehlein, J.; Hymes, S.R. Pustular psoriasis induced by infliximab. J. Drugs Dermatol. 2004, 3, 439–440. [Google Scholar] [PubMed]
- Beuthien, W.; Mellinghoff, H.-U.; Von Kempis, J. Skin reaction to adalimumab. Arthritis Rheum. 2004, 50, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Dereure, O.; Guillot, B.; Jorgensen, C.; Cohen, J.D.; Combes, B.; Guilhou, J.J. Psoriatic lesions induced by antitumour necrosis factor-alpha treatment: Two cases. Br. J. Dermatol. 2004, 151, 506–507. [Google Scholar] [CrossRef]
- Collamer, A.N.; Battafarano, D. Psoriatic Skin Lesions Induced by Tumor Necrosis Factor Antagonist Therapy: Clinical Features and Possible Immunopathogenesis. Semin. Arthritis Rheum. 2010, 40, 233–240. [Google Scholar] [CrossRef]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; DeMaria, O.; Navarini, A.A.; Lapointe, A.-K.; French, L.E.; Vernez, M.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef]
- Harrison, M.J.; Dixon, W.G.; Watson, K.D.; King, Y.; Groves, R.; Hyrich, K.L.; Symmons, D.P.M. Rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor α therapy: Results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2009, 68, 209–215. [Google Scholar] [CrossRef]
- George, L.A.; Gadani, A.; Cross, R.K.; Jambaulikar, G.; Ghazi, L.J. Psoriasiform Skin Lesions Are Caused by Anti-TNF Agents Used for the Treatment of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 3424–3430. [Google Scholar] [CrossRef]
- Weizman, A.V.; Sharma, R.; Afzal, N.M.; Xu, W.; Walsh, S.; Stempak, J.M.; Nguyen, G.C.; Croitoru, K.; Steinhart, A.H.; Silverberg, M.S. Stricturing and Fistulizing Crohn’s Disease Is Associated with Anti-tumor Necrosis Factor-Induced Psoriasis in Patients with Inflammatory Bowel Disease. Dig. Dis. Sci. 2018, 63, 2430–2438. [Google Scholar] [CrossRef]
- Palucka, A.K.; Blanck, J.-P.; Bennett, L.; Pascual, V.; Banchereau, J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 3372–3377. [Google Scholar] [CrossRef] [PubMed]
- De Gannes, G.C.; Ghoreishi, M.; Pope, J.; Russell, A.; Bell, D.; Adams, S.; Shojania, K.; Martinka, M.; Dutz, J.P. Psoriasis and pustular dermatitis triggered by TNF-{alpha} inhibitors in patients with rheumatologic conditions. Arch Dermatol. 2007, 143, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Papp, K.; Maari, C.; Yao, Y.; Robbie, G.; White, W.I.; Le, C.; White, B. A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J. Am. Acad. Dermatol. 2010, 62, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.C. Atopic dermatitis-like eruption precipitated by infliximab. J. Am. Acad. Dermatol. 2003, 49, 160–161. [Google Scholar] [CrossRef]
- Flendrie, M.; Vissers, W.H.; Creemers, M.C.; de Jong, E.M.; van de Kerkhof, P.C.; van Riel, P.L. Dermatological conditions during TNF-alpha-blocking therapy in patients with rheumatoid arthritis: A prospective study. Arthritis Res. Ther. 2005, 7, R666–R676. [Google Scholar] [CrossRef]
- Lee, H.-H.; Song, I.-H.; Friedrich, M.; Gauliard, A.; Detert, J.; Röwert, J.; Audring, H.; Kary, S.; Burmester, G.-R.; Sterry, W.; et al. Cutaneous side-effects in patients with rheumatic diseases during application of tumour necrosis factor-α antagonists. Br. J. Dermatol. 2007, 156, 486–491. [Google Scholar] [CrossRef]
- Nakamura, M.; Lee, K.; Singh, R.; Zhu, T.H.; Farahnik, B.; Abrouk, M.; Koo, J.; Bhutani, T. Eczema as an adverse effect of anti-TNFα therapy in psoriasis and other Th1-mediated diseases: A review. J. Dermatol. Treat. 2017, 28, 237–241. [Google Scholar] [CrossRef]
- Zaba, L.C.; Cardinale, I.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Novitskaya, I.; Khatcherian, A.; Bluth, M.J.; Lowes, M.A.; et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J. Exp. Med. 2007, 204, 3183–3194. [Google Scholar] [CrossRef]
- Quaglino, P.; Bergallo, M.; Ponti, R.; Barberio, E.; Cicchelli, S.; Buffa, E.; Comessatti, A.; Costa, C.; Terlizzi, M.; Astegiano, S.; et al. Th1, Th2, Th17 and Regulatory T Cell Pattern in Psoriatic Patients: Modulation of Cytokines and Gene Targets Induced by Etanercept Treatment and Correlation with Clinical Response. Dermatology 2011, 223, 57–67. [Google Scholar] [CrossRef]
- Malisiewicz, B.; Murer, C.; Schmid, J.P.; French, L.E.; Schmid-Grendelmeier, P.; Navarini, A.A. Eosinophilia during psoriasis treatment with TNF antagonists. Dermatology 2011, 223, 311–315. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Thomas, P.; Breit, S.; Dugas, M.; Mailhammer, R.; Van Eden, W.; Van Der Zee, R.; Biedermann, T.; Prinz, J.; Mack, M.; et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat. Med. 2003, 9, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.; Maier, H.; Riedl, E.; Brüggen, M.C.; Reininger, B.; Schaschinger, M.; Bangert, C.; Guenova, E.; Stingl, G.; Brunner, P.M. Analysis of anti-tumour necrosis factor-induced skin lesions reveals strong T helper 1 activation with some distinct immunological characteristics. Br. J. Dermatol. 2018, 178, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Ariyasu, T.; Tanaka, T.; Fujioka, N.; Yanai, Y.; Yamamoto, S.; Yamauchi, H.; Ikegami, H.; Ikeda, M.; Kurimoto, M. Effects of interferon-alpha subtypes on the TH1/TH2 balance in peripheral blood mononuclear cells from patients with hepatitis virus infection-associated liver disorders. In Vitro Cell. Dev. Biol. Anim. 2005, 41, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Esmailzadeh, A.; Yousefi, P.; Farhi, D.; Bachmeyer, C.; Cosnes, J.; Berenbaum, F.; Duriez, P.; Aractingi, S.; Khosrotehrani, K. Predictive Factors of Eczema-Like Eruptions among Patients without Cutaneous Psoriasis Receiving Infliximab: A Cohort Study of 92 Patients. Dermatology 2009, 219, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Takase, K.; Horton, S.C.; Ganesha, A.; Das, S.; McHugh, A.; Emery, P.; Savic, S.; Buch, M.H. What is the utility of routine ANA testing in predicting development of biological DMARD-induced lupus and vasculitis in patients with rheumatoid arthritis? Data from a single-centre cohort. Ann. Rheum. Dis. 2014, 73, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Benucci, M.; Saviola, G.; Baiardi, P.; Cammelli, E.; Manfredi, M. Anti-nucleosome antibodies as prediction factor of development of autoantibodies during therapy with three different TNFα blocking agents in rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 91–95. [Google Scholar] [CrossRef]
- Moulis, G.; Sommet, A.; Lapeyre-Mestre, M.; Montastruc, J.-L. Is the risk of tumour necrosis factor inhibitor-induced lupus or lupus-like syndrome the same with monoclonal antibodies and soluble receptor? A case/non-case study in a nationwide pharmacovigilance database. Rheumatology 2014, 53, 1864–1871. [Google Scholar] [CrossRef]
- Jani, M.; Dixon, W.G.; Kersley-Fleet, L.; Bruce, I.N.; Chinoy, H.; Barton, A.; Lunt, M.; Watson, K.; Symmons, D.P.; Hyrich, K.L.; et al. Drug-specific risk and characteristics of lupus and vasculitis-like events in patients with rheumatoid arthritis treated with TNFi: Results from BSRBR-RA. RMD Open 2017, 3, e000314. [Google Scholar] [CrossRef]
- Vedove, C.D.; Del Giglio, M.; Schena, D.; Girolomoni, G. Drug-induced lupus erythematosus. Arch. Dermatol. Res. 2009, 301, 99–105. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; Muñoz, S.; Soria, N.; Galiana, D.; Bertolaccini, L.; Maria-Jose, C.; Munther, A.K. Autoimmune diseases induced by TNF-targeted therapies: Analysis of 233 cases. Medicine 2007, 86, 242–251. [Google Scholar] [CrossRef]
- Jacob, C.O.; McDevitt, H.O. Tumour necrosis factor-α in murine autoimmune ‘lupus’ nephritis. Nature 1988, 331, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.O.; Fronek, Z.; Lewis, G.D.; Koo, M.; Hansen, J.A.; McDevitt, H.O. Heritable major histocompatibility complex class II-associated differences in production of tumor necrosis factor alpha: Relevance to genetic predisposition to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1990, 87, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Via, C.S.; Shustov, A.; Rus, V.; Lang, T.; Nguyen, P.; Finkelman, F.D. In vivo neutralization of TNF-alpha promotes humoral autoimmunity by preventing the induction of CTL. J. Immunol. 2001, 167, 6821–6826. [Google Scholar] [CrossRef] [PubMed]
- Karamanakos, A.; Vergou, T.; Panopoulos, S.; Tektonidou, M.G.; Stratigos, A.J.; Sfikakis, P.P. Psoriasis as an adverse reaction to biologic agents beyond anti-TNF-α therapy. Eur. J. Dermatol. 2021, 31, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Pernet, C.; Guillot, B.; Bessis, D. Eczematous Drug Eruption after Ustekinumab Treatment. Arch. Dermatol. 2012, 148, 959–960. [Google Scholar] [CrossRef]
- Al-Janabi, A.; Foulkes, A.; Mason, K.; Smith, C.; Griffiths, C.; Warren, R. Phenotypic switch to eczema in patients receiving biologics for plaque psoriasis: A systematic review. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1440–1448. [Google Scholar] [CrossRef]
- Ezzedine, K.; Visseaux, L.; Cadiot, G.; Brixi, H.; Bernard, P.; Reguiai, Z. Ustekinumab for skin reactions associated with anti-tumor necrosis factor-α agents in patients with inflammatory bowel diseases: A single-center retrospective study. J. Dermatol. 2019, 46, 322–327. [Google Scholar] [CrossRef]
- Wu, J.; Smogorzewski, J. Ustekinumab for the treatment of paradoxical skin reactions and cutaneous manifestations of inflammatory bowel diseases. Dermatol. Ther. 2021, 34, e14883. [Google Scholar] [CrossRef]
- Teraki, Y.; Takahashi, A.; Inoue, Y.; Takamura, S. Eyelid Dermatitis as a Side Effect of Interleukin-17A Inhibitors in Psoriasis. Acta Derm. Venereol. 2018, 98, 456–457. [Google Scholar] [CrossRef]
- Burlando, M.; Cozzani, E.; Russo, R.; Parodi, A. Atopic-like dermatitis after secukinumab injection: A case report. Dermatol. Ther. 2019, 32, e12751. [Google Scholar] [CrossRef]
- Napolitano, M.; Megna, M.; Fabbrocini, G.; Nisticò, S.; Balato, N.; Dastoli, S.; Patruno, C. Eczematous eruption during anti-interleukin 17 treatment of psoriasis: An emerging condition. Br. J. Dermatol. 2019, 181, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Caldarola, G.; Pirro, F.; Di Stefani, A.; Talamonti, M.; Galluzzo, M.; D’Adamio, S.; Magnano, M.; Bernardini, N.; Malagoli, P.; Bardazzi, F.; et al. Clinical and histopathological characterization of eczematous eruptions occurring in course of anti IL-17 treatment: A case series and review of the literature. Expert Opin. Biol. Ther. 2020, 20, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Danset, M.; Hacard, F.; Jaulent, C.; Nosbaum, A.; Berard, F.; Nicolas, J.-F.; Goujon, C. Brodalumab-associated generalized eczematous eruption in a difficult-to-treat psoriasis patient: Management without brodalumab withdrawal. Eur. J. Dermatol. 2020, 30, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Brembilla, N.C.; Senra, L.; Boehncke, W.-H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef]
- Crowley, J.; Warren, R.; Cather, J. Safety of selective IL -23p19 inhibitors for the treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1676–1684. [Google Scholar] [CrossRef]
- Reyn, B.; Gils, A.; Hillary, T. Eczematous eruption after guselkumab treatment for psoriasis. JAAD Case Rep. 2019, 5, 973–975. [Google Scholar] [CrossRef]
- Truong, A.; Le, S.; Kiuru, M.; Maverakis, E. Nummular dermatitis on guselkumab for palmoplantar psoriasis. Dermatol. Ther. 2019, 32, e12954. [Google Scholar] [CrossRef]
- Miyagawa, F.; Fukuda, K.; Mori, A.; Ogawa, K.; Asada, H. Recurrence of secukinumab-induced eczematous eruptions after guselkumab treatment for pustular psoriasis. J. Dermatol. 2021, 48, E498–E499. [Google Scholar] [CrossRef]
- Kromer, C.; Schön, M.P.; Mössner, R. Eczematous eruption in patients with psoriasis during risankizumab treatment. Eur. J. Dermatol. 2020, 30, 599–601. [Google Scholar] [CrossRef]
- Narla, S.; Silverberg, J.I.; Simpson, E.L. Management of inadequate response and adverse effects to dupilumab in atopic dermatitis. J. Am. Acad. Dermatol. 2022, 86, 628–636. [Google Scholar] [CrossRef]
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab Treatment in Adults with Moderate-to-Severe Atopic Dermatitis. N. Engl. J. Med. 2014, 371, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.-P.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Tracey, E.H.; Elston, C.; Feasel, P.; Piliang, M.; Michael, M.; Vij, A. Erythrodermic presentation of psoriasis in a patient treated with dupilumab. JAAD Case Rep. 2018, 4, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Safa, G.; Paumier, V. Psoriasis induced by dupilumab therapy. Clin. Exp. Dermatol. 2019, 44, e49–e50. [Google Scholar] [CrossRef]
- Gori, N.; Caldarola, G.; Pirro, F.; De Simone, C.; Peris, K. A case of guttate psoriasis during treatment with dupilumab. Dermatol. Ther. 2019, 32, e12998. [Google Scholar] [CrossRef]
- Stout, M.; Guitart, J.; Tan, T.; Silverberg, J.I. Psoriasis-like Dermatitis Developing in a Patient with Atopic Dermatitis Treated with Dupilumab. Dermatitis 2019, 30, 376–378. [Google Scholar] [CrossRef]
- Napolitano, M.; Scalvenzi, M.; Fabbrocini, G.; Cinelli, E.; Patruno, C. Occurrence of psoriasiform eruption during dupilumab therapy for adult atopic dermatitis: A case series. Dermatol. Ther. 2019, 32, e13142. [Google Scholar] [CrossRef]
- Napolitano, M.; Ferrillo, M.; Patruno, C.; Scalvenzi, M.; D’Andrea, M.; Fabbrocini, G. Efficacy and Safety of Dupilumab in Clinical Practice: One Year of Experience on 165 Adult Patients from a Tertiary Referral Centre. Dermatol. Ther. 2021, 11, 355–361. [Google Scholar] [CrossRef]
- Jaulent, L.; Staumont-Sallé, D.; Tauber, M.; Paul, C.; Aubert, H.; Marchetti, A.; Sassolas, B.; Valois, A.; Nicolas, J.; Nosbaum, A.; et al. De novo psoriasis in atopic dermatitis patients treated with dupilumab: A retrospective cohort. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e296–e297. [Google Scholar] [CrossRef]
- Brumfiel, C.M.; Patel, M.H.; Zirwas, M.J. Development of psoriasis during treatment with dupilumab: A systematic review. J. Am. Acad. Dermatol. 2022, 86, 708–709. [Google Scholar] [CrossRef]
- Guenova, E.; Skabytska, Y.; Hoetzenecker, W.; Weindl, G.; Sauer, K.; Tham, M.; Kim, K.W.; Park, J.H.; Seo, J.H.; Ignatova, D.; et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2015, 112, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Caiazzo, G.; Fabbrocini, G.; Balato, A.; Di Caprio, R.; Scala, E.; Scalvenzi, M.; Patruno, C. Increased expression of interleukin-23A in lesional skin of patients with atopic dermatitis with psoriasiform reaction during dupilumab treatment. Br. J. Dermatol. 2021, 184, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.; Wang, A.; Ramachandran, S.; Damsky, W.; Cohen, J. Dupilumab-induced phenotype switch from atopic dermatitis to psoriasis is characterized by de novo interleukin-17A expression: A case report. Br. J. Dermatol. 2021, 185, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Chen, J.K.; Chiou, A.; Ko, J.; Honari, G. Assessment of the Development of New Regional Dermatoses in Patients Treated for Atopic Dermatitis with Dupilumab. JAMA Dermatol. 2019, 155, 850–852. [Google Scholar] [CrossRef]
- Soria, A.; Du-Thanh, A.; Seneschal, J.; Jachiet, M.; Staumont-Sallé, D.; Barbarot, S.; GREAT Research Group. Development or Exacerbation of Head and Neck Dermatitis in Patients Treated for Atopic Dermatitis with Dupilumab. JAMA Dermatol. 2019, 155, 1312–1315. [Google Scholar] [CrossRef]
- Buhl, T.; Sulk, M.; Nowak, P.; Buddenkotte, J.; McDonald, I.; Aubert, J.; Carlavan, I.; Deret, S.; Reiniche, P.; Rivier, M.; et al. Molecular and Morphological Characterization of Inflammatory Infiltrate in Rosacea Reveals Activation of Th1/Th17 Pathways. J. Investig. Dermatol. 2015, 135, 2198–2208. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Indications | Class | Medication | Description |
|---|---|---|---|
| Psoriasis | TNF-α inhibitors | Infliximab | chimeric mAb to TNF-α |
| Adalimumab | fully human mAb to TNF-α | ||
| Certolizumab pegol | humanized mAb to TNF-α | ||
| Etanercept | recombinant human TNF-R/IgGFc | ||
| IL-17 inhibitors | Secukinumab | fully human mAb to IL-17A | |
| Brodalumab | fully human mAb to IL-17RA | ||
| Ixekizumab | humanized mAb to IL-17A | ||
| IL-12/23p40 inhibitor | Ustekinumab | fully human mAb to IL-12/23p40 | |
| IL-23p19 inhibitors | Guselkumab | fully human mAb to IL-23p19 | |
| Risankizumab | humanized mAb to IL-23p19 | ||
| Tildrakizumab | humanized mAb to IL-23p19 | ||
| AD | IL-4Rα inhibitor | Dupilumab | fully human mAb to IL-4Rα |
| Pt No. | Author | Age | Sex | Clinical Description (Duration) | Biologic | Time of Onset | Previous Atopy | Histology | Clinical Course |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Reyn et al. (2019) | 47 | M | Psoriasis vulgaris (NR) | Guselkumab | 10 w | AD | Acanthosis, spongiosis, lymphocytic inflammatory infiltrates mixed with eosinophils | Guselkumab discontinued; Eczema resolved with tar preparation |
| 2 | Truong et al. (2019) | 40 | M | Pustular psoriasis (since childhood) | Guselkumab | 3 m | NR | Psoriasiform epidermal hyperplasia, parakeratosis, spongiosis, perivascular lymphohistiocytic infiltrates | NR |
| 3 | Miyagawa et al. (2021) | 75 | M | Pustular psoriasis (13 years) | Guselkumab | 3 m | None | Parakeratosis, spongiosis, perivascular inflammatory infiltrates consisting of lymphocytes and eosinophils | Switched from secukinumab due to eczematous eruptions; Guselkumab continued; Treated with topical corticosteroids; Eczema persisted |
| 4 | Kromer et al. (2020) | 52 | M | Psoriasis vulgaris (4 years) | Risankizumab | 3 w | Allergic rhinitis | Acanthosis, compact orthokeratosis with foci of parakeratosis, spongiosis, perivascular lymphocytic infiltrates | Guselkumab continued; Improved after treatment with topical corticosteroids |
| 5 | as above | 59 | M | Psoriasis vulgaris (26 years) | Risankizumab | 4 w | Allergic rhinitis, Asthma | NR | Switched to ustekinumab; Eczema improved |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyagawa, F. Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines 2022, 10, 1485. https://doi.org/10.3390/biomedicines10071485
Miyagawa F. Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines. 2022; 10(7):1485. https://doi.org/10.3390/biomedicines10071485
Chicago/Turabian StyleMiyagawa, Fumi. 2022. "Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases" Biomedicines 10, no. 7: 1485. https://doi.org/10.3390/biomedicines10071485
APA StyleMiyagawa, F. (2022). Pathogenesis of Paradoxical Reactions Associated with Targeted Biologic Agents for Inflammatory Skin Diseases. Biomedicines, 10(7), 1485. https://doi.org/10.3390/biomedicines10071485