
An Oral 3D Printed PLGA-Tocopherol PEG Succinate Nanocomposite Hydrogel for High-Dose Methotrexate Delivery in Maintenance Chemotherapy

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Phase 1: Synthesis of Methotrexate-Loaded Tocopheryl Polyethylene Glycol Succinate-Functionalized Polylactide-Co-Glycolic Acid Nanoparticles
2.2.1. Preparation of Methotrexate-Loaded TPGS-PLGA Nanoparticulate System
2.2.2. Componential Analysis of Chemical Structure Integrity Post Nanoparticle Formation
2.2.3. Nanometric Characterization of the MTX-Loaded TPGS-Functionalized PLGA Nanoparticles
2.2.4. Morphology Characterization of MTX-Loaded TPGS-PLGA Nanoparticles
2.2.5. Thermal Analysis of Methotrexate-Loaded TPGS and PLGA Nanoparticles
2.2.6. In Vitro Release Evaluation of Methotrexate from the TPGS-PLGA Nanoparticles
2.3. In Silico Analysis of P-gp Inhibition by TPGS
2.4. Phase 2: Synthesis and Optimization of the Sodium Alginate-Gelatine Bio-Ink as a 3D Printable Matrix for Nanoparticle Fixation
2.4.1. Optimization of Printing Parameters
2.4.2. Printability and Optimization of Sodium Alginate Gelatine Bio-Ink
2.4.3. Optimum Hydrogel Printable Ink of Sodium Alginate/Gelatine
2.4.4. Determination of the Effect of Needle Gauge on 3D Printing Accuracy
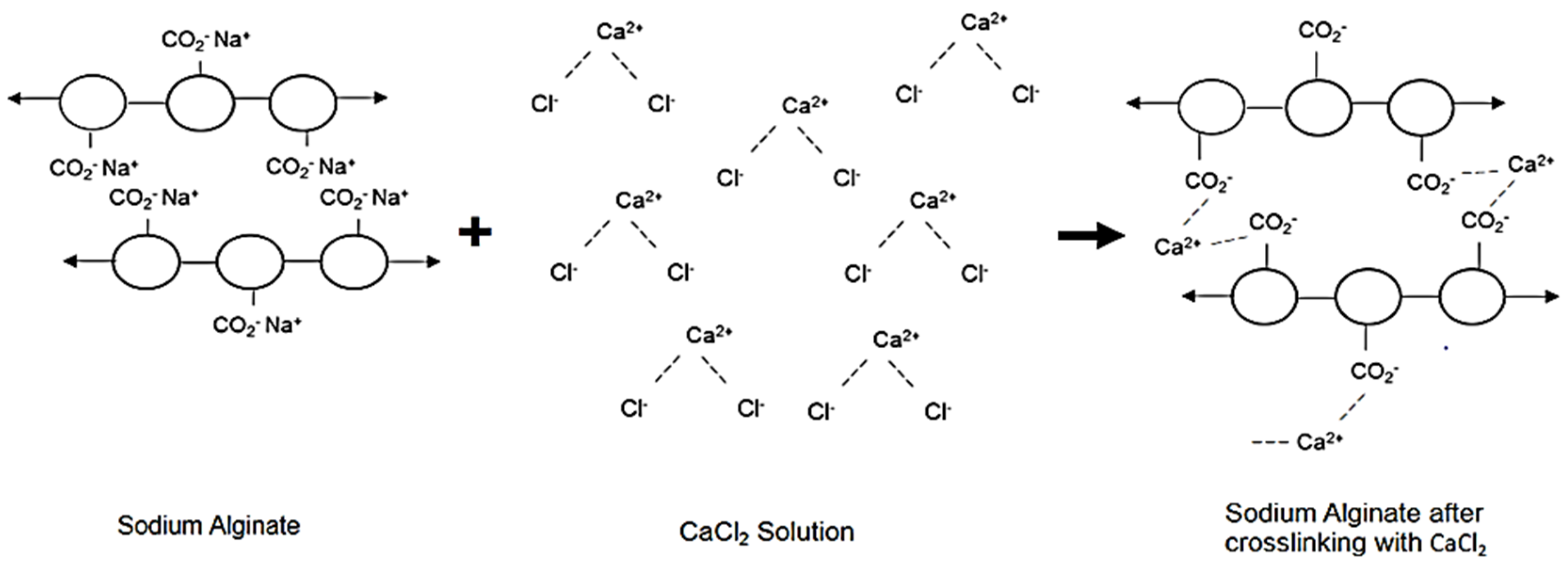
2.4.5. Crosslinking of Sodium Alginate
2.4.6. Temperature-Induced Crosslinking of Gelatine
2.4.7. Determination of Optimum Printing Speed and Pre- and Post-Flow Delay Times
2.5. Phase 3: Design and Synthesis of an Oral Chemotherapeutic 3D Printed Nanocomposite Hydrogel Tablet
2.5.1. Preparation of Sodium Alginate-Gelatine Nanoparticle Formulation Printing Ink
2.5.2. Design of 3D Printed Tablets
2.5.3. Thermal Analysis of 3D Printed Tablets
2.5.4. In Vitro Analysis of the 3D Printed Sodium Alginate-Gelatine Hydrogel-Nanoparticle Formulation for Oral Drug Delivery
3. Results and Discussion
3.1. Phase 1: Synthesis of Methotrexate-Loaded Tocopheryl Polyethylene Glycol Succinate-Functionalized Polylactide-Co-Glycolic Acid Nanoparticles
3.1.1. Assessment of MTX-Loaded TPGS-Functionalized PLGA Nanoparticles
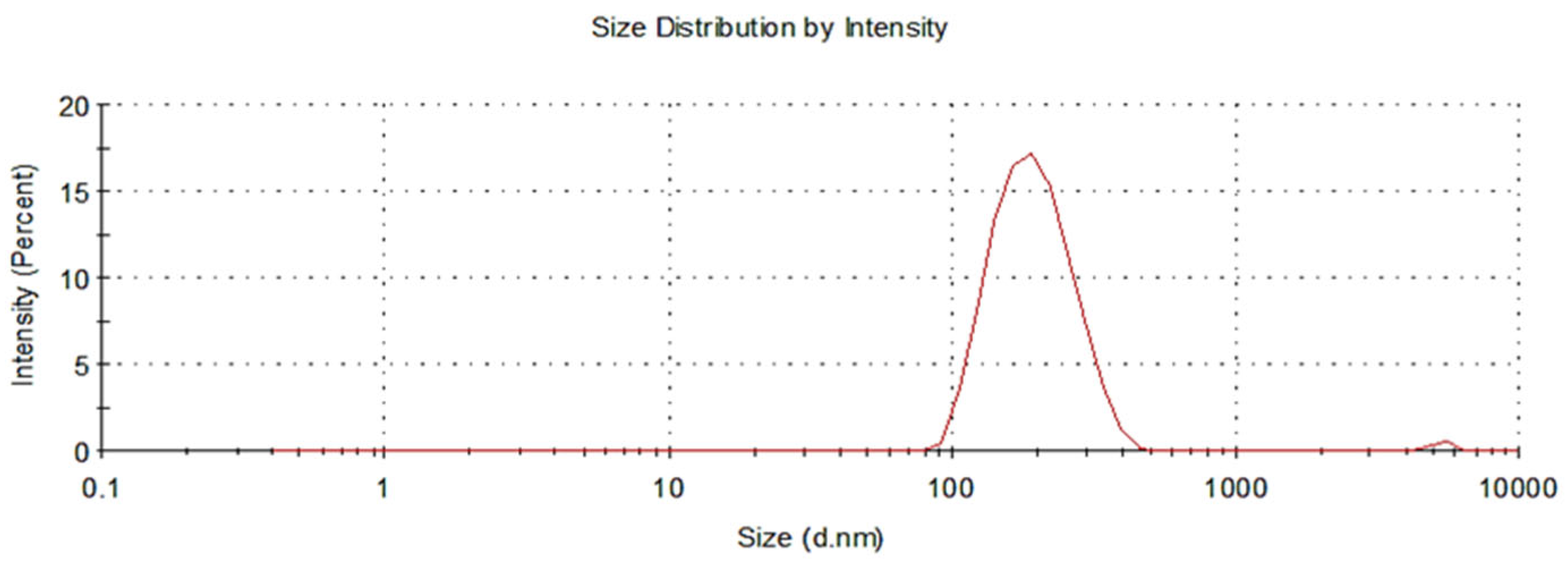
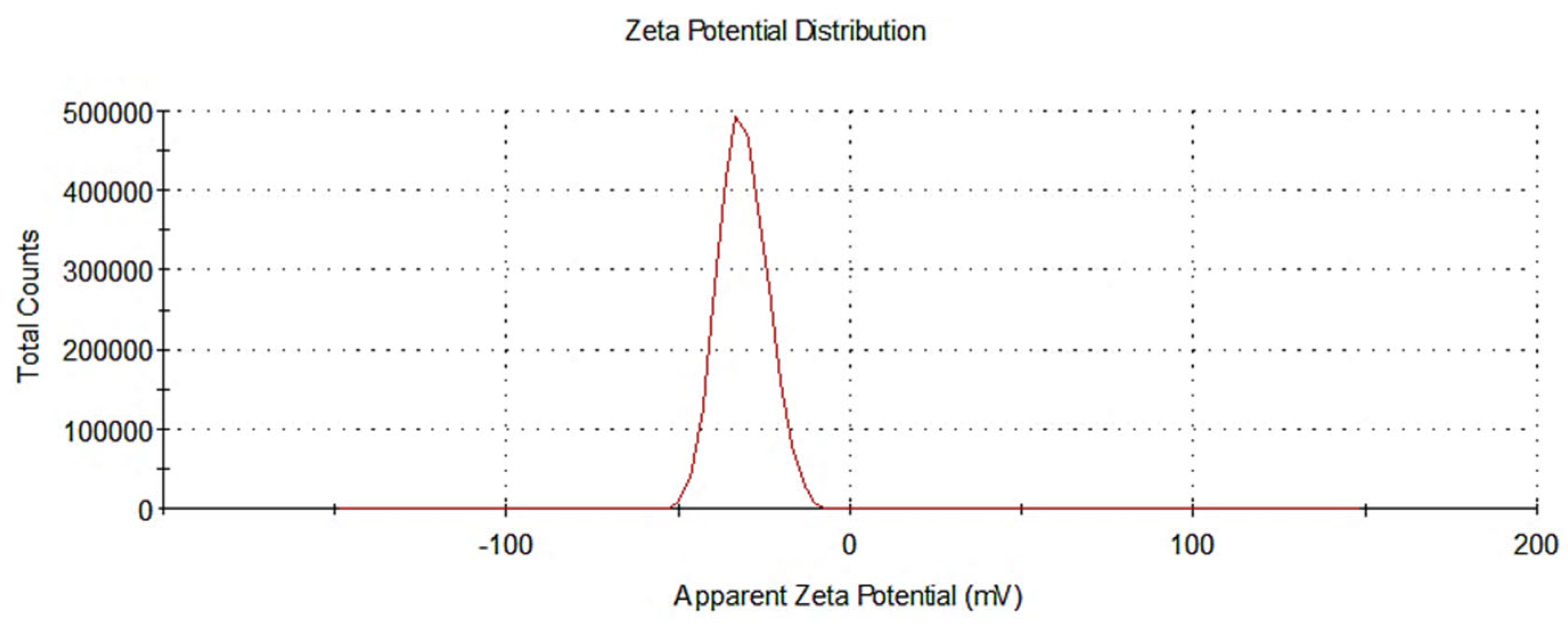
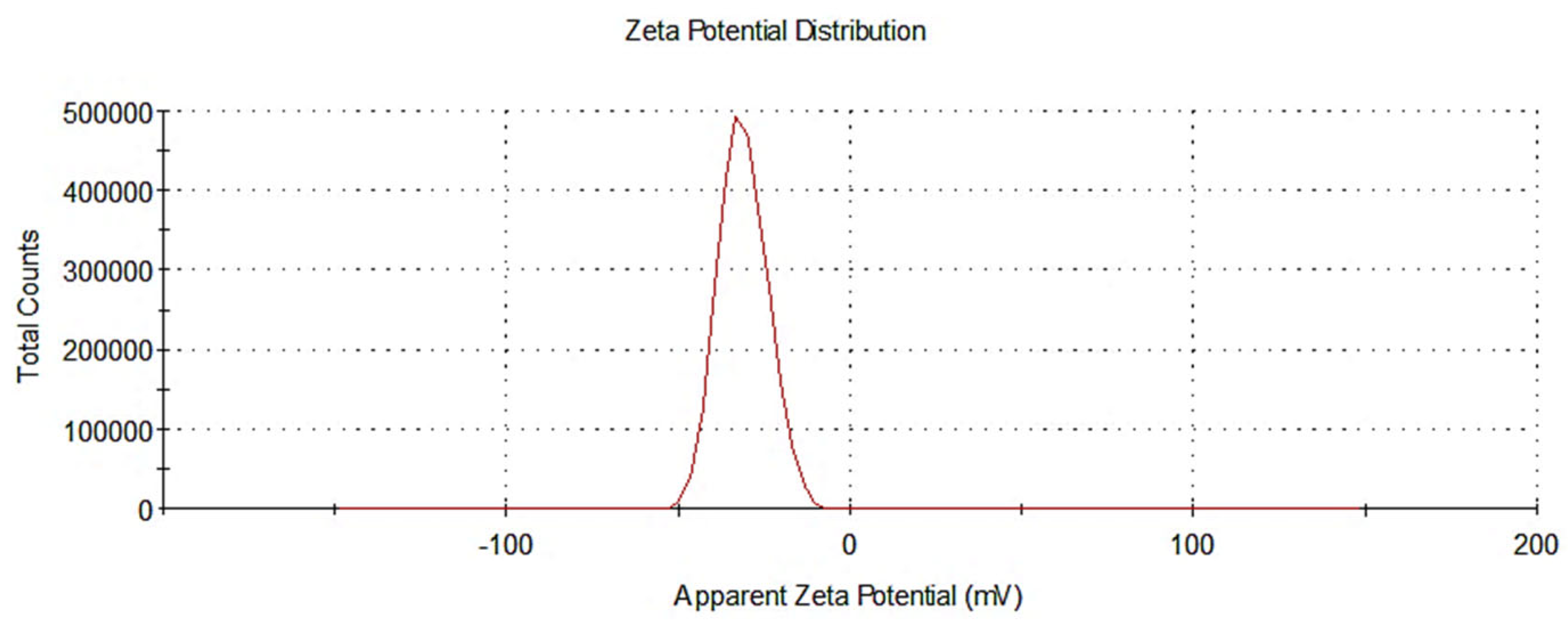
3.1.2. Assessment of Nanoparticle Size and Zeta Potential
3.1.3. Assessment of Nanoparticle Morphology
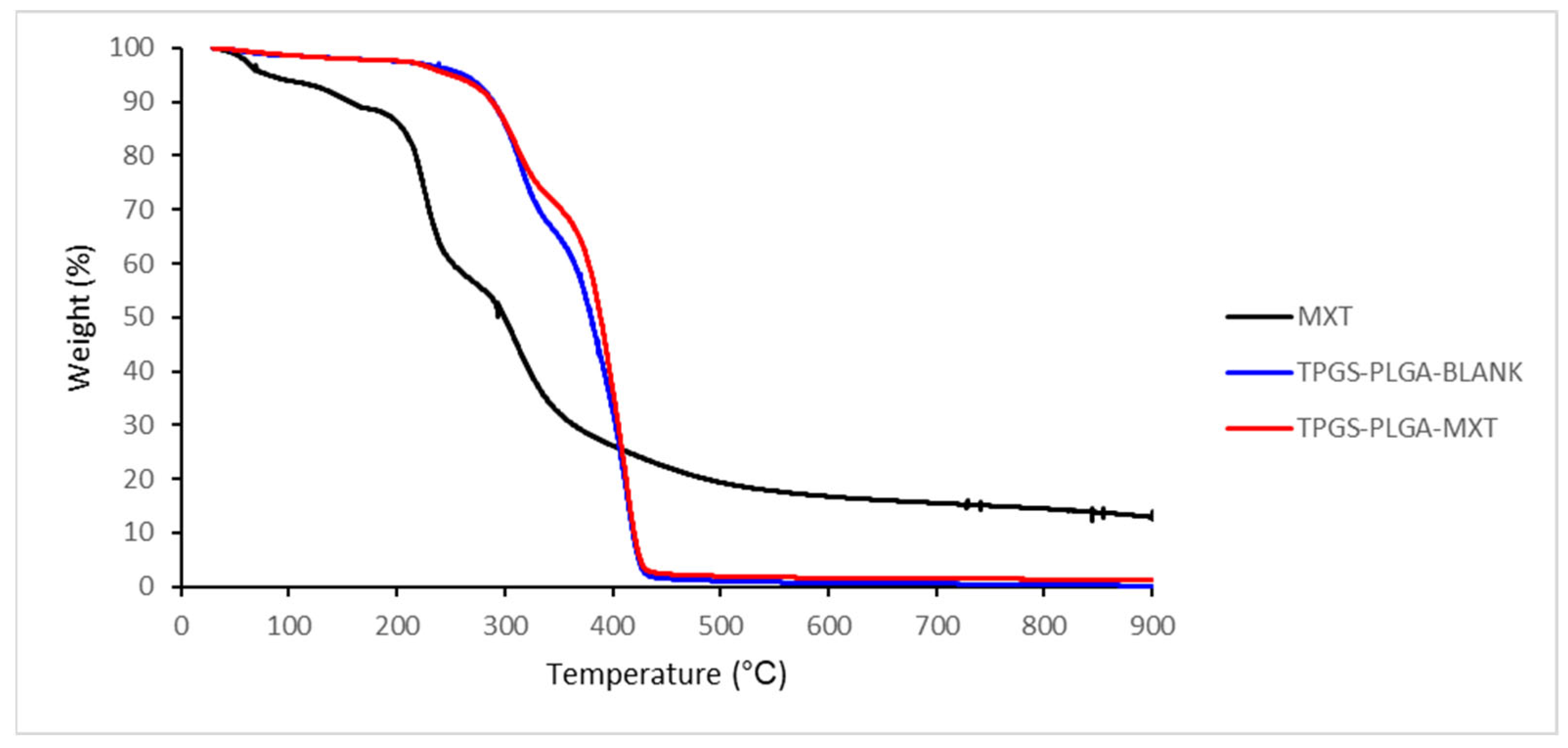
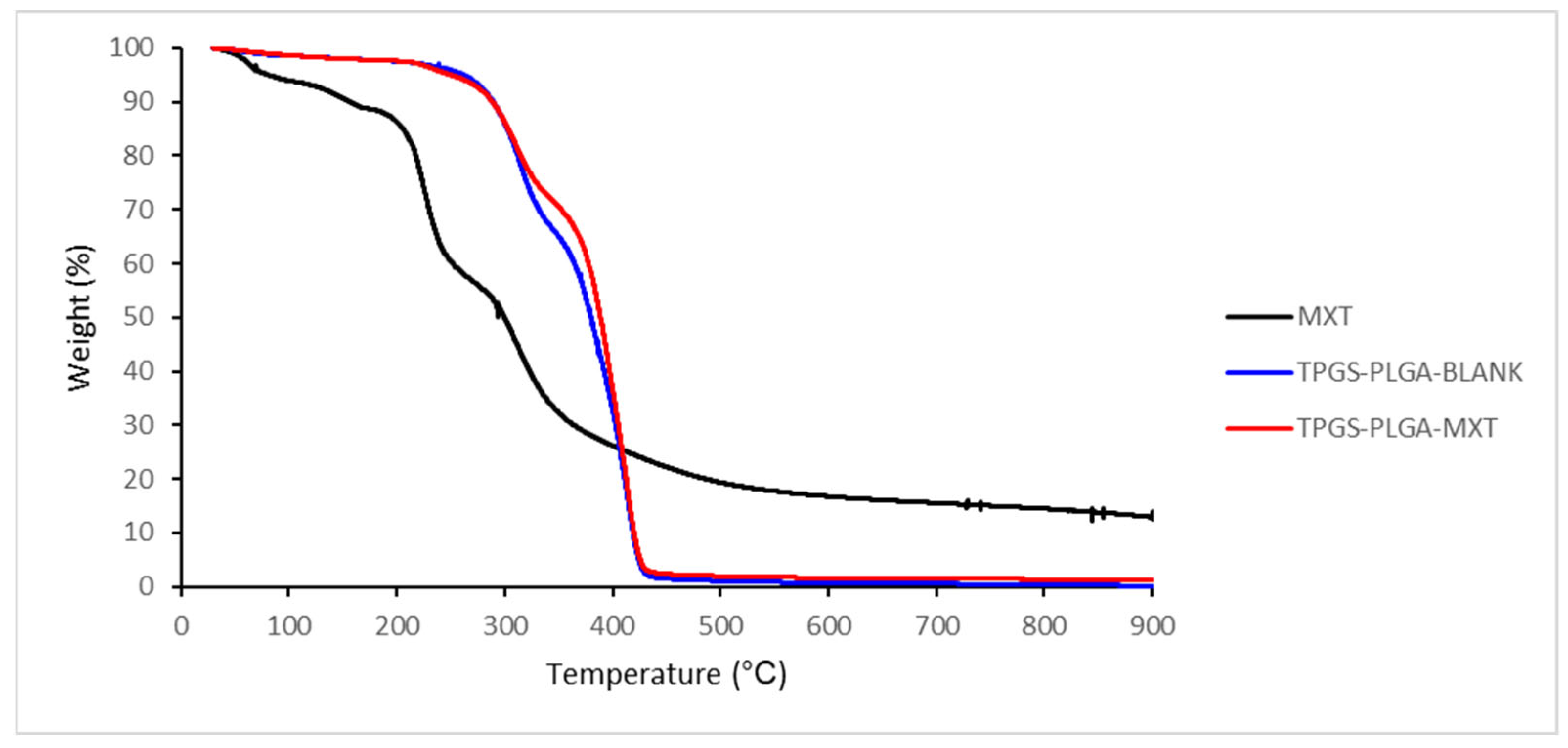
3.1.4. Thermogravimetric Analysis of MTX-Loaded TPGS-Functionalized PLGA Nanoparticles
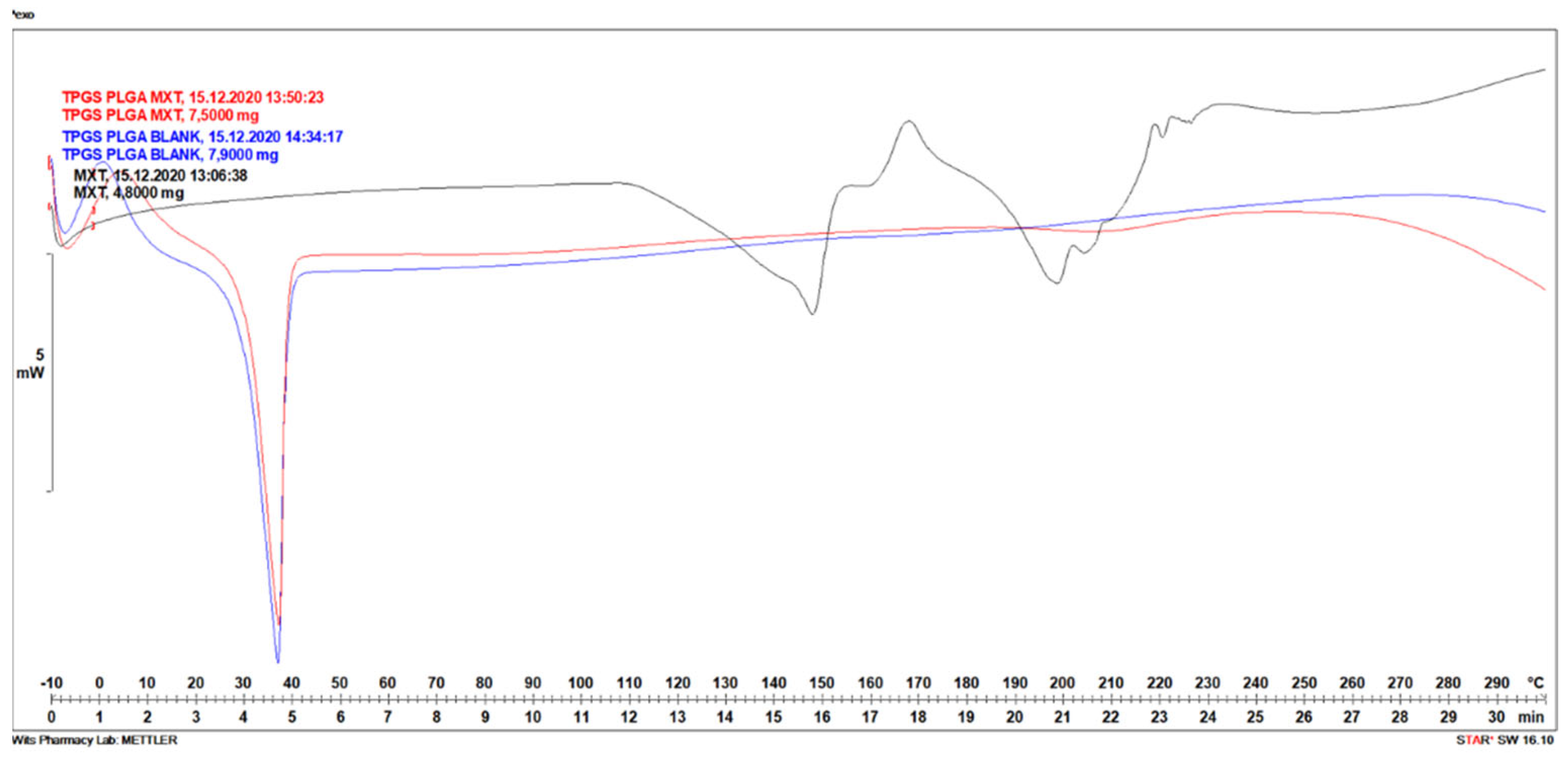
3.1.5. Differential Scanning Calorimetry Analysis of MTX-Loaded TPGS-Functionalized PLGA Nanoparticles
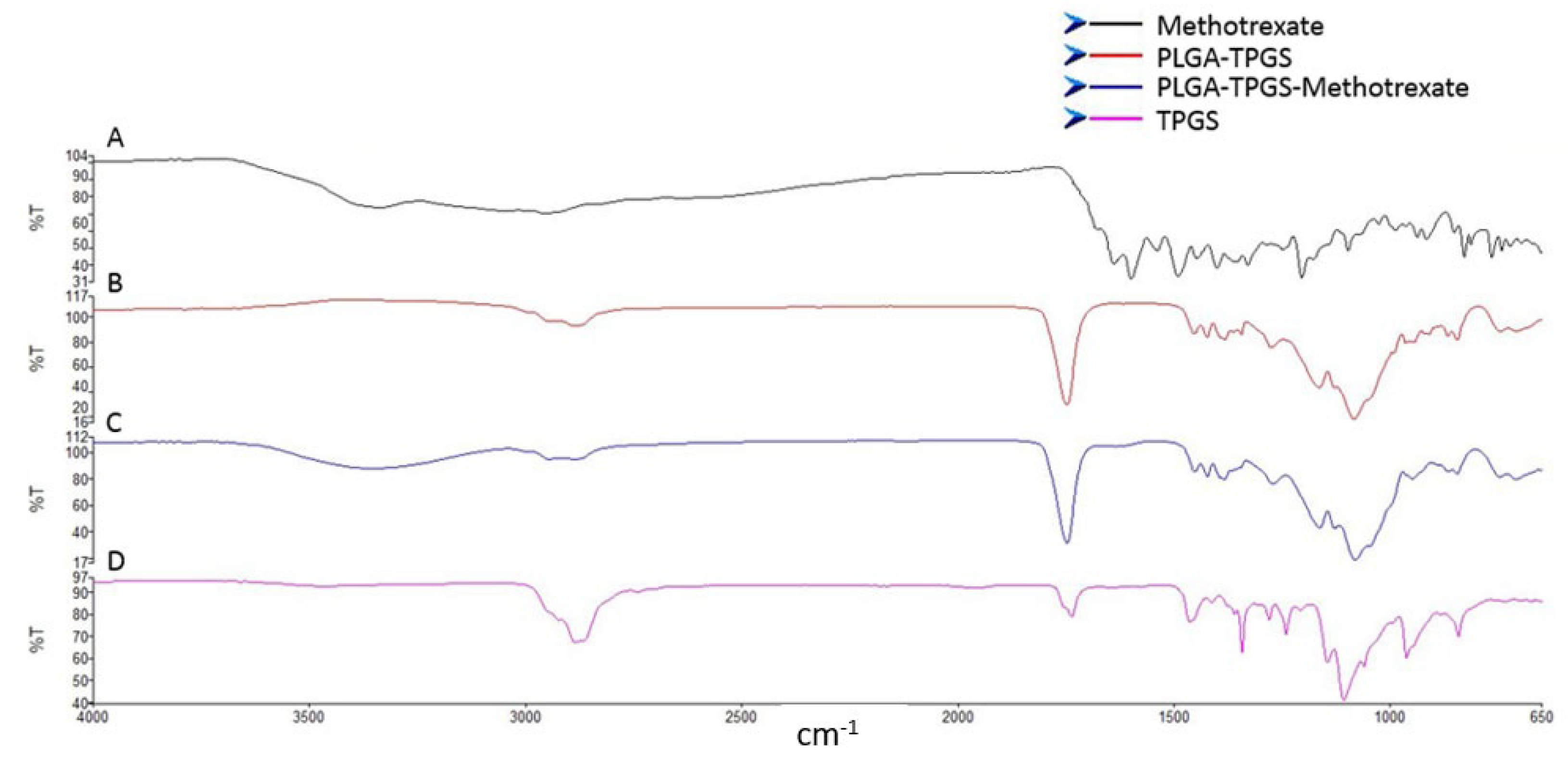
3.1.6. Evaluation of the Chemical Stability and Integrity of the Various Nanosystem Components
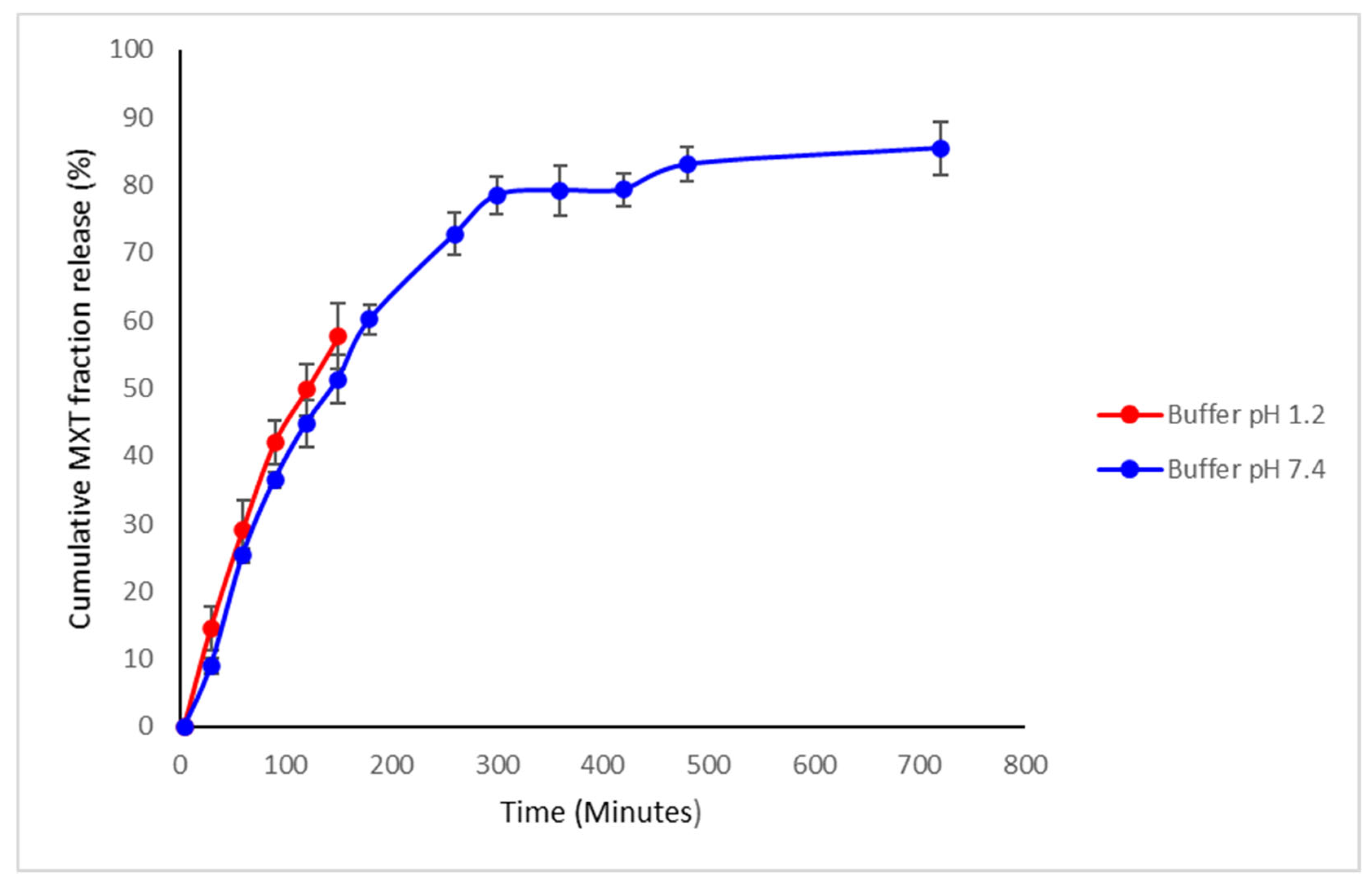
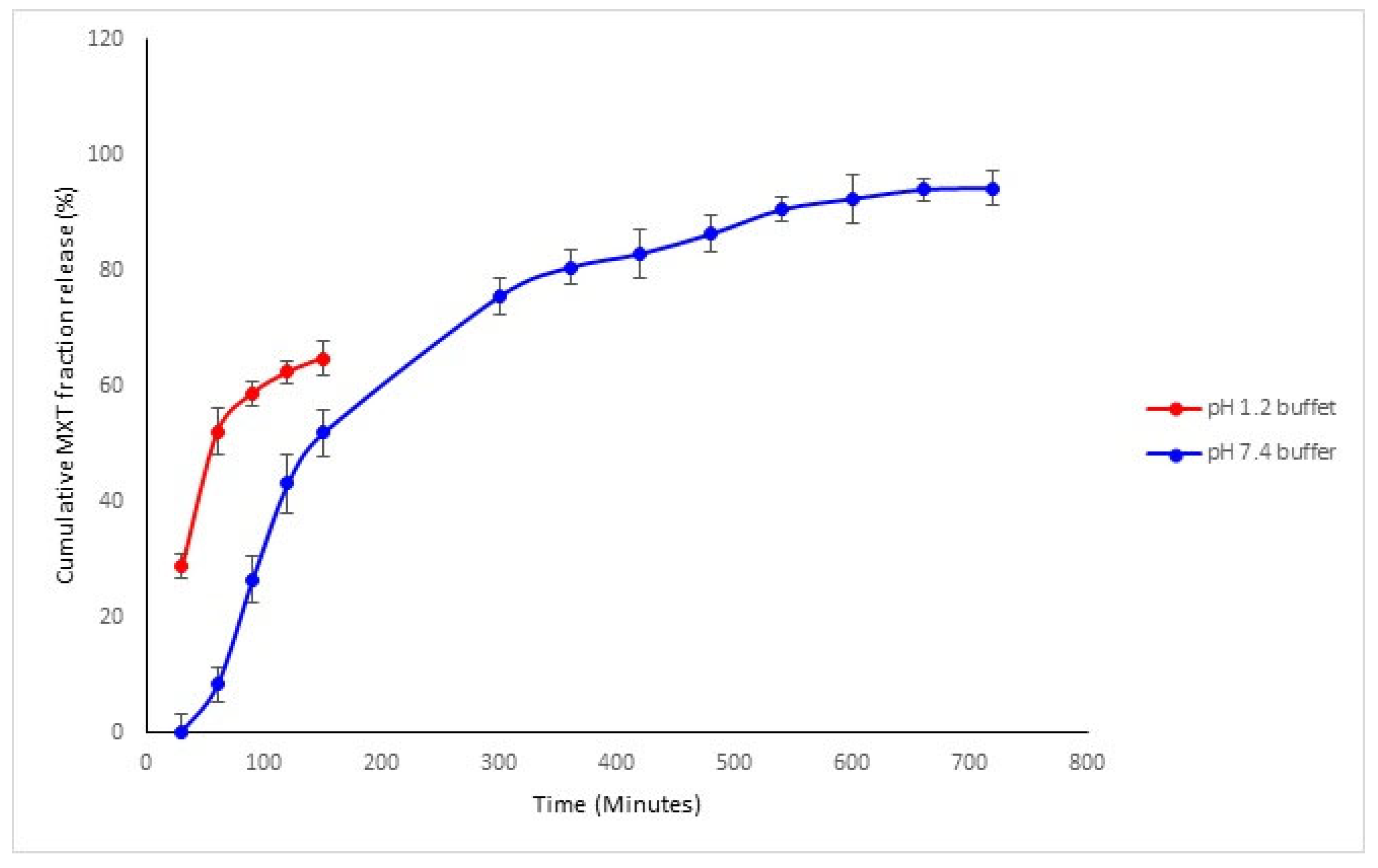
3.1.7. Drug Release Kinetics of Methotrexate from TPGS and PLGA Nanoparticles
3.2. Phase 2: Synthesis and Optimization of the Sodium Alginate-Gelatine Bio-Ink as a 3D Printable Matrix for Nanoparticle Fixation
3.2.1. Optimization of Sodium Alginate/Gelatine Printing Ink
3.2.2. The Influence of Printing Needle Size on 3D Printing Accuracy
3.2.3. Optimization of Crosslinking Time of Sodium Alginate-Gelatine Devices in CaCl2
3.2.4. Determination of Optimum Printing Temperature
3.2.5. The Influence of Printing Speed and Pre- and Post-Flow Dwell Times on 3D Printing Accuracy
3.2.6. The Influence of Temperature on 3D Printing Accuracy
3.2.7. Optimization of the Ionic Crosslinking Time of SA-GL Matrices
3.3. Phase 3: Design and Synthesis of an Oral Chemotherapeutic 3D Printed Nanocomposite Hydrogel Tablet
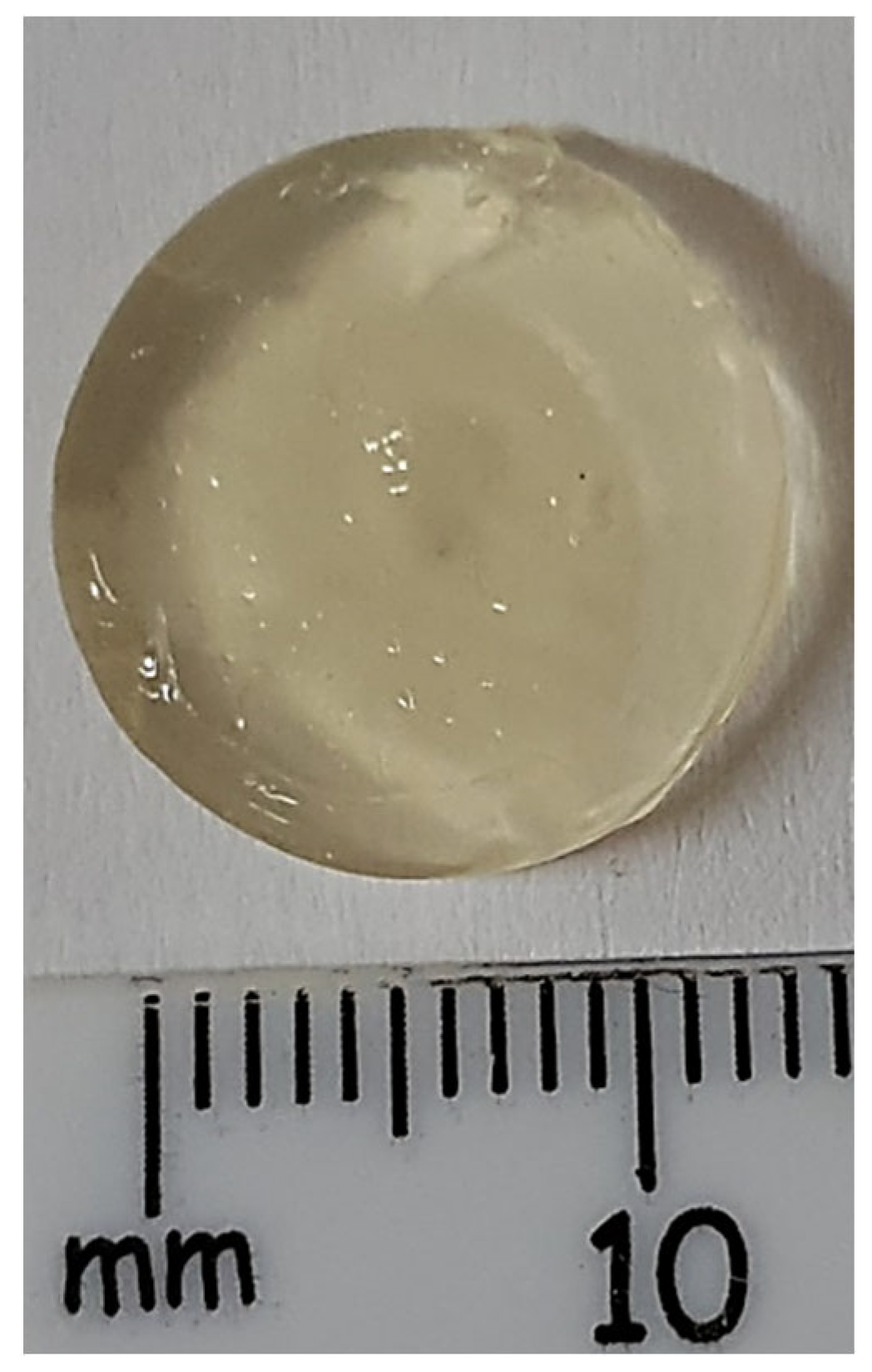
3.3.1. The 3D Printing of Sodium Alginate-Gelatine Hydrogel Nanoparticle Formulation (Oral Chemotherapeutic Delivery System)
3.3.2. Determination of Thermal Stability of the Sodium Alginate-Gelatine Nanoparticle Formulation
3.3.3. Drug Release Profiles of the Sodium Alginate-Gelatine Hydrogel Nanoparticulate System
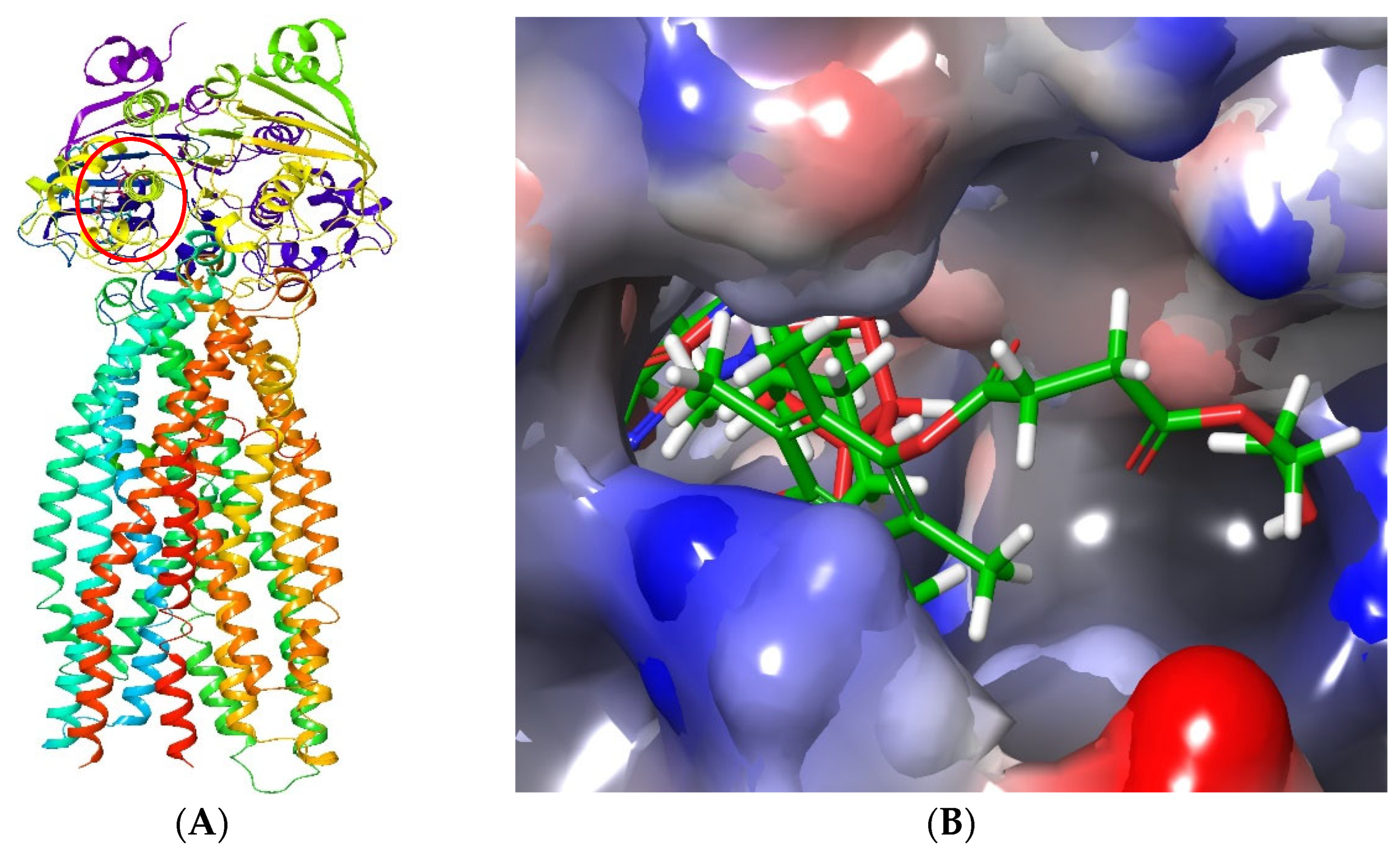
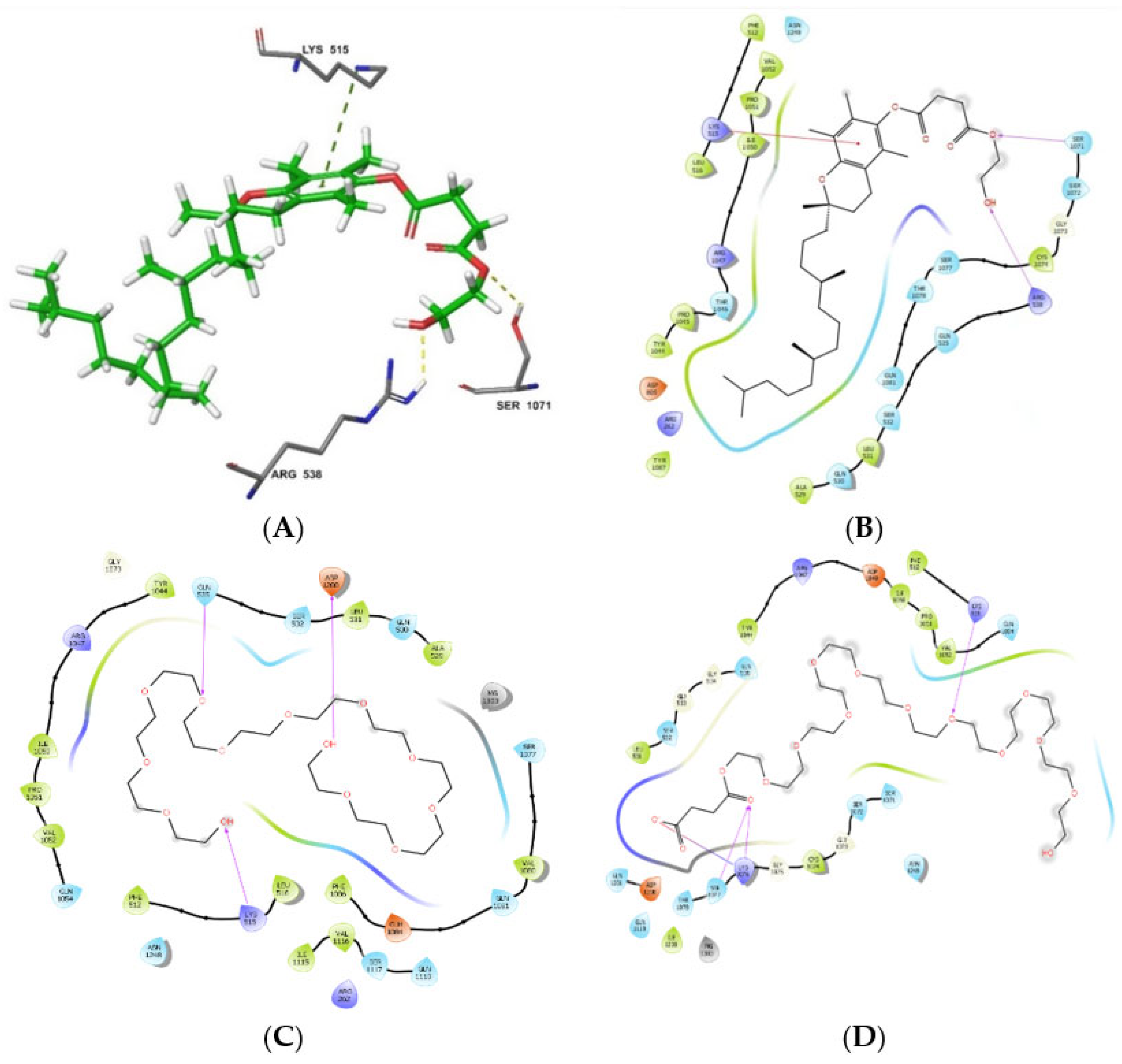
3.4. Molecular Modelling of P-gp Inhibition by the TPGS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chem. Phar. 2013, 71, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, Y.; Li, R.; Wang, T.; Han, M.; Zhu, C.; Wang, X. Methotrexate nanoparticles prepared with codendrimer from polyamidoamine (PAMAM) and oligoethylene glycols (OEG) dendrons: Antitumor efficacy in vitro and in vivo. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qian, X.W.; Zhu, X.H.; Yu, Y.; Miao, H.; Meng, J.H.; Jiang, J.Y.; Wang, H.S.; Zhai, X.W. Population pharmacokinetics of high-dose methotrexate in Chinese pediatric patients with acute lymphoblastic leukemia. Front. Pharmacol. 2021, 12, 1791. [Google Scholar] [CrossRef]
- Bleyer, W.A. The clinical pharmacology of methotrexate. New applications of an old drug. Cancer 1978, 41, 36–51. [Google Scholar] [PubMed]
- Grim, J.; Chládek, J.; Martínková, J. Pharmacokinetics and pharmacodynamics of methotrexate in non-neoplastic diseases. Clin. Pharm. 2003, 42, 139–151. [Google Scholar] [CrossRef]
- Khan, Z.A.; Tripathi, R.; Mishra, B. Methotrexate: A detailed review on drug delivery and clinical aspects. Expert Opin. Drug Deliv. 2012, 9, 151–169. [Google Scholar] [CrossRef]
- Walvekar, P.; Gannimani, R.; Govender, T. Combination drug therapy via nanocarriers against infectious diseases. Eur. J. Pharm. Sci. 2019, 127, 121–141. [Google Scholar] [CrossRef]
- Teresi, M.E.; Crom, W.R.; Choi, K.E.; Mirro, J.; Evans, W.E. Methotrexate bioavailability after oral and intramuscular administration in children. J. Pediatr. 1987, 110, 788–792. [Google Scholar] [CrossRef]
- Amani, A.; Begdelo, J.M.; Yaghoubi, H.; Motallebinia, S. Multifunctional magnetic nanoparticles for controlled release of anticancer drug, breast cancer cell targeting, MRI/fluorescence imaging, and anticancer drug delivery. J. Drug Del. Sci. Technol. 2019, 49, 534–546. [Google Scholar] [CrossRef]
- Sanità, G.; Carrese, B.; Lamberti, A. Nanoparticle Surface Functionalization: How to Improve Biocompatibility and Cellular Internalization. Front. Mol. Biosci. 2020, 7, 381. [Google Scholar] [CrossRef]
- Jain, A.K.; Swarnakar, N.K.; Godugu, C.; Singh, R.P.; Jain, S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials 2011, 32, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.P.; Date, A.A.; Patravale, V.B. Overcoming poor oral bioavailability using nanoparticle formulations–opportunities and limitations. Drug Discov. Today Technol. 2012, 9, e87–e95. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Roy, I. Doxorubicin-loaded casein nanoparticles for drug delivery: Preparation, characterization and in vitro evaluation. Intl. J. Biol. Macromol. 2019, 121, 6–12. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of efflux pumps in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, L.Y.; Wang, B.; Ding, M.H.; Bao, Y.L.; Tan, S.W. Recent Advances of D-α-tocopherol Polyethylene Glycol 1000 Succinate Based Stimuli-responsive Nanomedicine for Cancer Treatment. Med. Sci. 2020, 40, 218–231. [Google Scholar] [CrossRef]
- Tan, S.; Zou, C.; Zhang, W.; Yin, M.; Gao, X.; Tang, Q. Recent developments in d-α-tocopheryl polyethylene glycol-succinate-based nanomedicine for cancer therapy. Drug Deliv. 2017, 24, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wu, T.; Qi, Y.; Zhang, Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics 2018, 8, 464. [Google Scholar] [CrossRef]
- Saneja, A.; Khare, V.; Alam, N.; Dubey, R.D.; Gupta, P.N. Advances in P-glycoprotein-based approaches for delivering anticancer drugs: Pharmacokinetic perspective and clinical relevance. Expert Opin. Drug Deliv. 2014, 11, 121–138. [Google Scholar] [CrossRef]
- Essa, D.; Kondiah, P.P.; Choonara, Y.E.; Pillay, V. The design of poly (lactide-co-glycolide) nanocarriers for medical applications. Front. Bioeng. Biotechnol. 2020, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, H.; Dorkoosh, F.; Adabi, M.; Khosravani, M.; Arzani, H.; Kamali, M. Methotrexate-loaded plga nanoparticles: Preparation, characterization and their cytotoxicity effect on human glioblastoma U87MG cells. Int. J. Med. Nano Res. 2017, 4, 020. [Google Scholar]
- Tan, K.X.; Danquah, M.K.; Sidhu, A.; Ongkudon, C.M.; Lau, S.Y. Towards targeted cancer therapy: Aptamer or oncolytic virus? Eur. J. Pharm. Sci. 2017, 96, 8–19. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Chung, T.S.; Ng, N.P. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials 2001, 22, 231–241. [Google Scholar] [CrossRef]
- Lu, B.; Lv, X.; Le, Y. Chitosan-modified PLGA nanoparticles for control-released drug delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Moroz, E.; Matoori, S.; Leroux, J.-C. Oral delivery of macromolecular drugs: Where we are after almost 100 years of attempts. Adv. Drug Deliv. Rev. 2016, 101, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Cerchiara, T.; Abruzzo, A.; Parolin, C.; Vitali, B.; Bigucci, F.; Gallucci, M.C.; Nicoletta, F.P.; Luppi, B. Microparticles based on chitosan/carboxymethylcellulose polyelectrolyte complexes for colon delivery of vancomycin. Carbohydr. Polym. 2016, 143, 124–130. [Google Scholar] [CrossRef]
- Rizwan, M.; Yahya, R.; Hassan, A.; Yar, M.; Azzahari, A.D.; Selvanathan, V.; Sonsudin, F.; Abouloula, C.N. pH sensitive hydrogels in drug delivery: Brief history, properties, swelling, and release mechanism, material selection and applications. Polymers 2017, 9, 137. [Google Scholar] [CrossRef]
- Ferreira, N.N.; Perez, T.A.; Pedreiro, L.N.; Prezotti, F.G.; Boni, F.I.; Cardoso, V.M.D.O.; Venâncio, T.; Gremião, M.P.D. A novel pH-responsive hydrogel-based on calcium alginate engineered by the previous formation of polyelectrolyte complexes (PECs) intended to vaginal administration. Drug Dev. Ind. Pharm. 2017, 43, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Mikolaszek, B.; Kazlauske, J.; Larsson, A.; Sznitowska, M. Controlled Drug Release by the Pore Structure in Polydimethylsiloxane Transdermal Patches. Polymers 2020, 12, 1520. [Google Scholar] [CrossRef] [PubMed]
- Schütz, K.; Placht, A.M.; Paul, B.; Brüggemeier, S.; Gelinsky, M.; Lode, A. Three-dimensional plotting of a cell-laden alginate/methylcellulose blend: Towards biofabrication of tissue engineering constructs with clinically relevant dimensions. J. Tissue Eng. Regen. Med. 2017, 11, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, K.; Isreb, A.; Alhnan, M.A. A flexible-dose dispenser for immediate and extended release 3D printed tablets. Eur. J. Pharm. Biopharm. 2015, 96, 380–387. [Google Scholar] [CrossRef]
- Shin, J.H.; Kang, H.-W. The development of gelatin-based bio-ink for use in 3D hybrid bioprinting. Int. J. Precis Eng. Manuf. 2018, 19, 767–771. [Google Scholar] [CrossRef]
- Wang, X.; Ao, Q.; Tian, X.; Fan, J.; Tong, H.; Hou, W.; Bai, S. Gelatin-based hydrogels for organ 3D bioprinting. Polymers 2017, 9, 401. [Google Scholar] [CrossRef] [Green Version]
- Isoda, T.; Ito, S.; Kajiwara, M.; Nagasawa, M. Successful high-dose methotrexate chemotherapy in a patient with acute lymphocytic leukemia who developed acute renal failure during the initial treatment. Pediatr. Int. 2007, 49, 1018–1019. [Google Scholar] [CrossRef]
- Cowan, D.S.; Tannock, I.F. Factors that influence the penetration of methotrexate through solid tissue. Int. J. Cancer 2001, 91, 120–125. [Google Scholar] [CrossRef]
- Alkholief, M.; Albasit, H.; Alhowyan, A.; Alshehri, S.; Raish, M.; Kalam, M.A.; Alshamsan, A. Employing a PLGA-TPGS based nanoparticle to improve the ocular delivery of Acyclovir. Saudi Pharm. J. 2019, 27, 293–302. [Google Scholar] [CrossRef]
- Gaonkar, R.H.; Ganguly, S.; Dewanjee, S.; Sinha, S.; Gupta, A.; Ganguly, S.; Chattopadhyay, D.; Chatterjee Debnath, M. Garcinol loaded vitamin E TPGS emulsified PLGA nanoparticles: Preparation, physicochemical characterization, in vitro and in vivo studies. Sci. Rep. 2017, 7, 1–14. [Google Scholar]
- Sardo, H.S.; Saremnejad, F.; Bagheri, S.; Akhgari, A.; Garekani, H.A.; Sadeghi, F. A review on 5-aminosalicylic acid colon-targeted oral drug delivery systems. Int. J. Pharm. 2019, 558, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, B.B.S.; Lasham, A.; Shelling, A.N.; Al-Kassas, R. Development of biodegradable PLGA nanoparticles surface engineered with hyaluronic acid for targeted delivery of paclitaxel to triple negative breast cancer cells. Mater. Sci. Eng. C 2017, 76, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Lawson, G.; Ogwu, J.; Tanna, S. Quantitative screening of the pharmaceutical ingredient for the rapid identification of substandard and falsified medicines using reflectance infrared spectroscopy. PLoS ONE 2018, 13, e0202059. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.; Jepson, M.A.; Tsuruo, T.; Simmons, N.L.; Hirst, B.H. Functional expression of P-glycoprotein in apical membranes of human intestinal Caco-2 cells. Kinetics of vinblastine secretion and interaction with modulators. J. Biol. Chem. 1993, 268, 14991–14997. [Google Scholar] [PubMed]
- Yano, K.; Tomono, T.; Ogihara, T. Advances in studies of P-glycoprotein and its expression regulators. Biol. Pharm. Bull. 2018, 41, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdzik, M.; Czekawy, I.; Oswald, S.; Drozdzik, A. Intestinal drug transporters in pathological states: An overview. Pharmacol. Rep. 2020, 72, 1173–1194. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, R.; Sangamwar, A.T. Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics. Biochim. Biophys. Acta (BBA)—Biomembr. 2014, 1838, 2882–2898. [Google Scholar] [CrossRef] [Green Version]
- Collnot, E.M.; Baldes, C.; Wempe, M.F.; Kappl, R.; Hüttermann, J.; Hyatt, J.A.; Edgar, K.J.; Schaefer, U.F.; Lehr, C.M. Mechanism of Inhibition of P-Glycoprotein Mediated Efflux by Vitamin E TPGS: Influence on ATPase Activity and Membrane Fluidity. Mol. Pharm. 2007, 4, 465–474. [Google Scholar] [CrossRef]
- Liu, T.; Liu, X.; Xiong, H.; Xu, C.; Yao, J.; Zhu, X.; Zhou, J.; Yao, J. Mechanisms of TPGS and its derivatives inhibiting P-glycoprotein efflux pump and application for reversing multidrug resistance in hepatocellular carcinoma. Polym. Chem. 2018, 9, 1827–1839. [Google Scholar] [CrossRef]
- Bogman, K.; Erne-Brand, F.; Alsenz, J.; Drewe, J. The role of surfactants in the reversal of active transport mediated by multidrug resistance proteins. J. Pharm. Sci. 2003, 92, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Luiz, M.T.; Di Filippo, L.D.; Alves, R.C.; Araújo, V.H.S.; Duarte, J.L.; Marchetti, J.M.; Chorilli, M. The use of TPGS in drug delivery systems to overcome biological barriers. Eur. Polym. J. 2021, 142, 110129. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.; Smolinski, M.P. Discovery and characterization of potent dual P-glycoprotein and CYP3A4 inhibitors: Design, synthesis, cryo-EM analysis, and biological evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Yu, H.; Zhang, L.; Hou, T. Computational models for predicting substrates or inhibitors of P-glycoprotein. Drug Discov. Today 2012, 17, 343–351. [Google Scholar] [CrossRef]
- Shahbaaz, M.; Nkaule, A.; Christoffels, A. Designing novel possible kinase inhibitor derivatives as therapeutics against Mycobacterium tuberculosis: An in silico study. Sci. Rep. 2019, 9, 4405. [Google Scholar] [CrossRef]
- Kryscio, D.R.; Shi, Y.; Ren, P.; Peppas, N.A. Molecular docking simulations for macromolecularly imprinted polymers. Ind. Eng. Chem. Res. 2011, 50, 13877–13884. [Google Scholar] [CrossRef] [Green Version]
- Crouch, E.C.; Smith, K.; McDonald, B.; Briner, D.; Linders, B.; McDonald, J.; Holmskov, U.; Head, J.; Hartshorn, K. Species differences in the carbohydrate binding preferences of surfactant protein D. Am. J. Respir. Cell Mol. Biol. 2006, 35, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Dolghih, E.; Bryant, C.; Renslo, A.R.; Jacobson, M.P. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comput. Biol. 2011, 7, e1002083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rants’o, T.A.; van der Westhuizen, C.J.; van Zyl, R.L. Optimization of covalent docking for organophosphates interaction with Anopheles acetylcholinesterase. J. Mol. Graph. Model. 2022, 110, 108054. [Google Scholar] [CrossRef] [PubMed]
- Fonkwe, L.G.; Narsimhan, G.; Cha, A.S. Characterization of gelation time and texture of gelatin and gelatin–polysaccharide mixed gels. Food Hydrocoll. 2003, 17, 871–883. [Google Scholar] [CrossRef]
- Park, H.E.; Gasek, N.; Hwang, J.; Weiss, D.J.; Lee, P.C. Effect of temperature on gelation and cross-linking of gelatin methacryloyl for biomedical applications. Phys. Fluids 2020, 32, 033102. [Google Scholar]
- Wan, X.; Woods, A.T.; Salgado-Montejo, A.; Velasco, C.; Spence, C. Assessing the expectations associated with pharmaceutical pill colour and shape. Food Qual. Prefer. 2015, 45, 171–182. [Google Scholar] [CrossRef]
- Lajoinie, A.; Henin, E.; Nguyen, K.A.; Malik, S.; Mimouni, Y.; Sapori, J.M.; Bréant, V.; Cochat, P.; Kassai, B. Oral drug dosage forms administered to hospitalized children: Analysis of 117,665 oral administrations in a French paediatric hospital over a 1-year period. Int. J. Pharm. 2016, 500, 336–344. [Google Scholar] [CrossRef]
- Ternik, R.; Liu, F.; Bartlett, J.A.; Khong, Y.M.; Tan, D.C.T.; Dixit, T.; Wang, S.; Galella, E.A.; Gao, Z.; Klein, S. Assessment of swallowability and palatability of oral dosage forms in children: Report from an M-CERSI pediatric formulation workshop. Int. J. Pharm. 2018, 536, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. [Google Scholar] [CrossRef]
- Shah, R.; Eldridge, D.; Palombo, E.; Harding, I. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J. Phys. Sci. 2014, 25, 73–91. [Google Scholar]
- Dixit, A.; Kulkarni, P.; Redyy, S. Methotrexate fast disintegrating tablet as a dosage form for dysphagia patients. Int. J. Pharm. Pharm. Sci. 2014, 6, 217–225. [Google Scholar]
- Pereira, A.D.F.; Pereira, L.G.R.; Barbosa, L.A.D.O.; Fialho, S.L.; Pereira, B.G.; Patricio, P.S.D.O.; Pinto, F.C.H.; Da Silva, G.R. Efficacy of methotrexate-loaded poly (ε-caprolactone) implants in Ehrlich solid tumor-bearing mice. Drug Deliv. 2013, 20, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuliaş, A.; Popoiu, C.; Vlase, G.; Vlase, T.; Oneţiu, D.; Săvoiu, G.; Simu, G.; Pătruţescu, C.; Ilia, G.; Ledeţi, I. Thermoanalytical and spectroscopic study on methotrexate–active substance and tablet. Dig. J. Nanomater. Biostruct 2014, 9, 93–98. [Google Scholar]
- Şimşek, S.; Eroğlu, H.; Kurum, B.; Ulubayram, K. Brain targeting of Atorvastatin loaded amphiphilic PLGA-b-PEG nanoparticles. J. Microencapsul. 2013, 30, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, M.; Law, N.; Webb, B.; Macrae, R.A.; Liew, L.J.; Sercombe, T.B.; Dilley, R.J.; Doyle, B.J. Mechanical behaviour of alginate-gelatin hydrogels for 3D bioprinting. J. Mech. Behav. Biomed. Mater. 2018, 79, 150–157. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Mao, S.; Sun, W.; Yao, R. The influence of printing parameters on cell survival rate and printability in microextrusion-based 3D cell printing technology. Biofabrication 2015, 7, 045002. [Google Scholar] [CrossRef]
- Xing, Q.; Yates, K.; Vogt, C.; Qian, Z.; Frost, M.C.; Zhao, F. Increasing mechanical strength of gelatin hydrogels by divalent metal ion removal. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Stodghill, S.P. THERMAL ANALYSIS: Thermal analysis—A review of techniques and applications in the pharmaceutical sciences. Am. Pharm. Rev. 2010, 13, 29. [Google Scholar]
- Ma, Y.; Zheng, Y.; Liu, K.; Tian, G.; Tian, Y.; Xu, L.; Yan, F.; Huang, L.; Mei, L. Nanoparticles of poly (lactide-co-glycolide)-da-tocopheryl polyethylene glycol 1000 succinate random copolymer for cancer treatment. Nanoscale Res. Lett. 2010, 5, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Kondu, S.; Ji, H.F.; McShane, M.J. Study of the near-neutral pH-sensitivity of chitosan/gelatin hydrogels by turbidimetry and microcantilever deflection. Biotechnol. Bioeng. 2006, 95, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, L.A.; Daily, A.M.; Horava, S.D.; Peppas, N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin. Drug Deliv. 2014, 11, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batrakova, E.V.; Li, S.; Li, Y.; Alakhov, V.Y.; Kabanov, A.V. Effect of pluronic P85 on ATPase activity of drug efflux transporters. Pharm. Res. 2004, 21, 2226–2233. [Google Scholar] [CrossRef] [PubMed]
- Collnot, E.M.; Baldes, C.; Schaefer, U.F.; Edgar, K.J.; Wempe, M.F.; Lehr, C.M. Vitamin E TPGS P-Glycoprotein Inhibition Mechanism: Influence on Conformational Flexibility, Intracellular ATP Levels, and Role of Time and Site of Access. Mol. Pharm. 2010, 7, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Charman, W.N.; Porter, C.J. An in vitro examination of the impact of polyethylene glycol 400, Pluronic P85, and vitamin E d-alpha-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSci 2002, 4, E40. [Google Scholar] [CrossRef]
- Shen, Q.; Lin, Y.; Handa, T.; Doi, M.; Sugie, M.; Wakayama, K.; Okada, N.; Fujita, T.; Yamamoto, A. Modulation of intestinal P-glycoprotein function by polyethylene glycols and their derivatives by in vitro transport and in situ absorption studies. Int. J. Pharm. 2006, 313, 49–56. [Google Scholar] [CrossRef]
- Hugger, E.D.; Novak, B.L.; Burton, P.S.; Audus, K.L.; Borchardt, R.T. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J. Pharm. Sci. 2002, 91, 1991–2002. [Google Scholar] [CrossRef]
- Gurjar, R.; Chan, C.Y.; Curley, P.; Sharp, J.; Chiong, J.; Rannard, S.; Siccardi, M.; Owen, A. Inhibitory effects of commonly used excipients on P-Glycoprotein in vitro. Mol. Pharm. 2018, 15, 4835–4842. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SA-GL Blend | Printing Pressure (kPa) | Matrix Size (mm2) | Printing Accuracy (%) |
|---|---|---|---|
| 3%SA-5%GL | 800 | 175.12 ± 15.03 | 77.15 ± 6.62 |
| 5%SA-5%GL | 800 | 197.74 ± 21.05 | 87.11 ± 9.27 |
| 5%SA-8%GL | 900 | 247.23 ± 24.82 | 91.09 ± 8.91 |
| 8%SA-10%GL | 1300 | 219.92 ± 5.44 | 96.88 ± 2.40 |
| 10%SA-10%GL | 1500 | 250.65 ± 6.35 | 89.58 ± 2.80 |
| Needle Gauge (G) | Needle Inner Diameter (mm) | Optimal Printing Pressure (kPa) | Printing Accuracy (%) |
|---|---|---|---|
| 20 | 0.61 | 100 | 73.3 |
| 21 | 0.51 | 300 | 76.7 |
| 22 | 0.41 | 800 | 87.9 |
| 25 | 0.25 | 1400 | 95.6 |
| 27 | 0.20 | 2000 | 82.3 |
| Extrusion at | 5 min | 10 min | 15 min | 20 min | 30 min | 45 min | 60 min | 90 min | 120 min |
|---|---|---|---|---|---|---|---|---|---|
| 30 °C | ✓ | ✓ | ✕ | ✕ | ✕ | ✕ | ✕ | ✕ | ✕ |
| 35 °C | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✕ | ✕ |
| 40 °C | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Printing Speed (mm/s) | Printed Matrix Size (mm2) | Printing Accuracy (%) | Visual Inspection |
|---|---|---|---|
| 25 | N/A | N/A | Bio-ink was viscous and L2 resulted in printing needle extrusion into L3. |
| 30 | 262 | 84 | Bio-ink was over-extruded |
| 35 | 227 | 99 | Matrix was well-formed with no structural defects |
| 40 | 212 | 94 | Matrix had several structural defects |
| 45 | N/A | N/A | Matrix had several structural defects and was not capable of L formation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondiah, P.P.D.; Rants’o, T.A.; Makhathini, S.S.; Mdanda, S.; Choonara, Y.E. An Oral 3D Printed PLGA-Tocopherol PEG Succinate Nanocomposite Hydrogel for High-Dose Methotrexate Delivery in Maintenance Chemotherapy. Biomedicines 2022, 10, 1470. https://doi.org/10.3390/biomedicines10071470
Kondiah PPD, Rants’o TA, Makhathini SS, Mdanda S, Choonara YE. An Oral 3D Printed PLGA-Tocopherol PEG Succinate Nanocomposite Hydrogel for High-Dose Methotrexate Delivery in Maintenance Chemotherapy. Biomedicines. 2022; 10(7):1470. https://doi.org/10.3390/biomedicines10071470
Chicago/Turabian StyleKondiah, Pierre P. D., Thankhoe A. Rants’o, Sifiso S. Makhathini, Sipho Mdanda, and Yahya E. Choonara. 2022. "An Oral 3D Printed PLGA-Tocopherol PEG Succinate Nanocomposite Hydrogel for High-Dose Methotrexate Delivery in Maintenance Chemotherapy" Biomedicines 10, no. 7: 1470. https://doi.org/10.3390/biomedicines10071470
APA StyleKondiah, P. P. D., Rants’o, T. A., Makhathini, S. S., Mdanda, S., & Choonara, Y. E. (2022). An Oral 3D Printed PLGA-Tocopherol PEG Succinate Nanocomposite Hydrogel for High-Dose Methotrexate Delivery in Maintenance Chemotherapy. Biomedicines, 10(7), 1470. https://doi.org/10.3390/biomedicines10071470