
Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Housing
2.2. Cloning
2.3. Viral Particles Production
2.4. In Vivo Virus Injection in the Sciatic Nerve
2.5. Sciatic Nerve Set Up under Multiphoton Microscope
2.6. Multiphoton Image Acquisition and Analysis
2.7. Immunohistochemistry
2.8. Intranerve Drug Administration
2.9. Systemic Drug Administration
2.10. CARS Imaging and Image Analysis
2.11. Cell Culture and Transfection
2.12. Protein Extraction and Western Blotting
2.13. Validation of Fluorescent Probes and Anesthesia Control
2.14. Electron Microscopy and Morphometry
2.15. Electrophysiology
2.16. Data and Statistical Analysis
3. Results
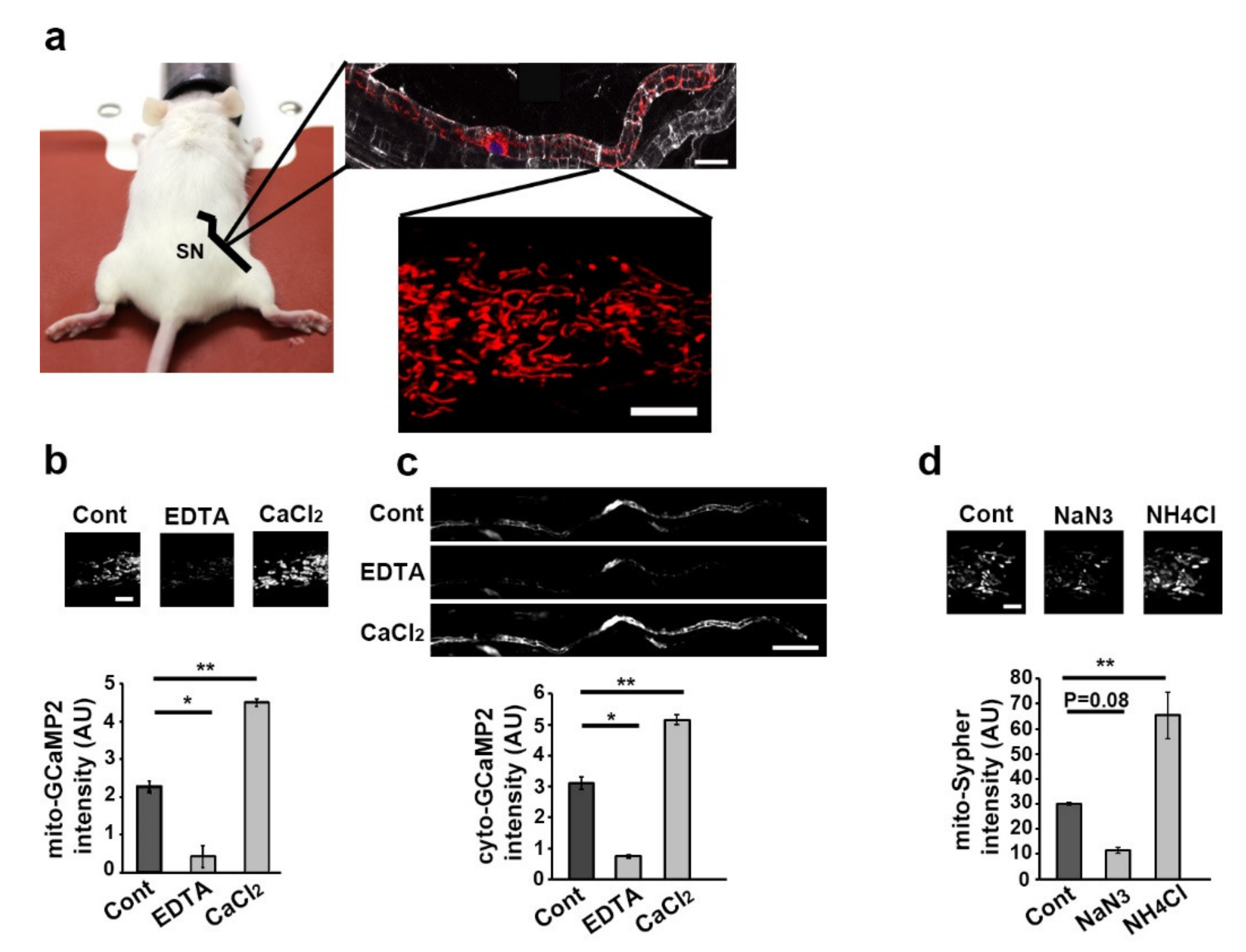
3.1. Live Imaging of mSC Mitochondria and Cytoplasm and Fluorescent Probes Validation
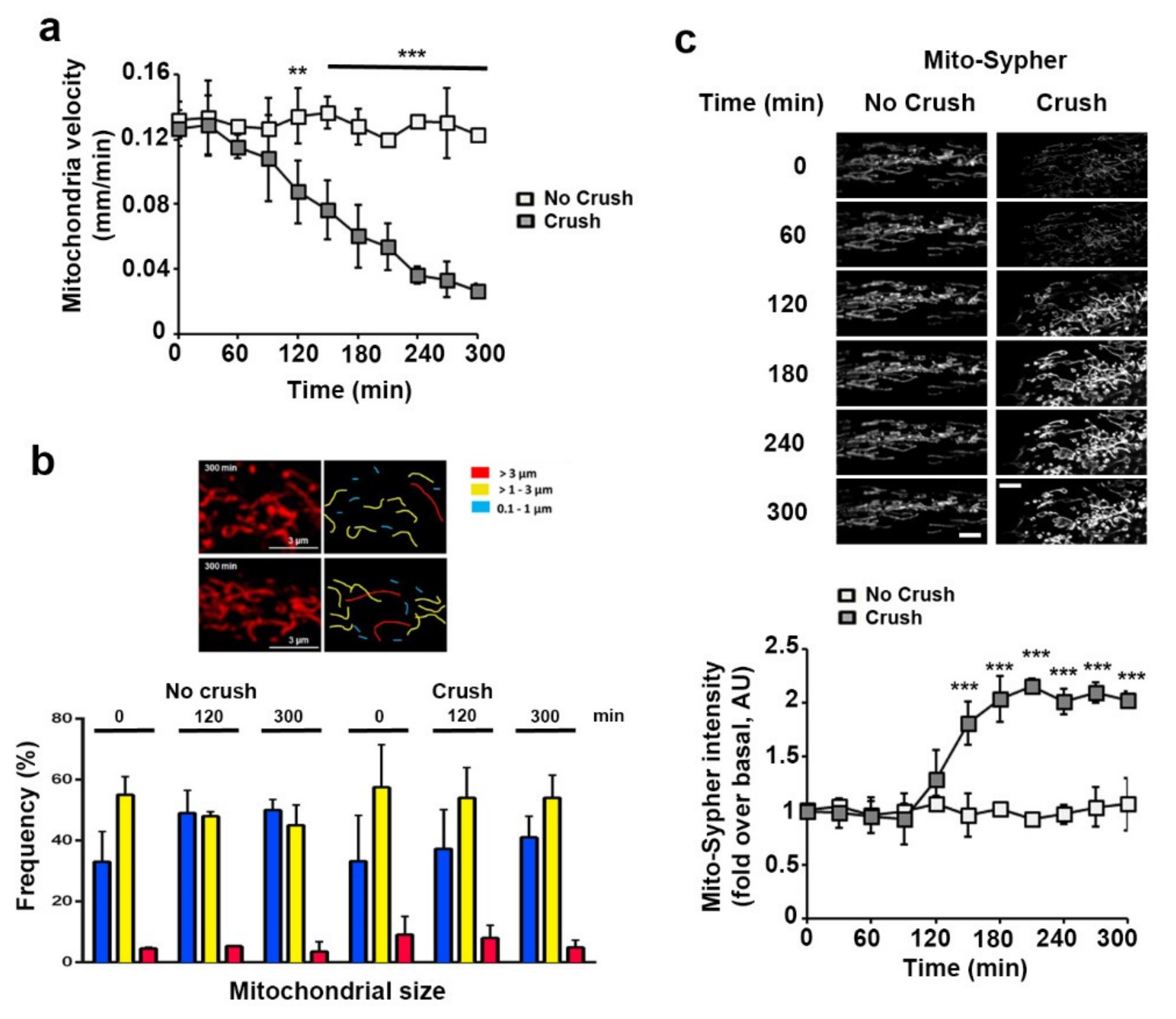
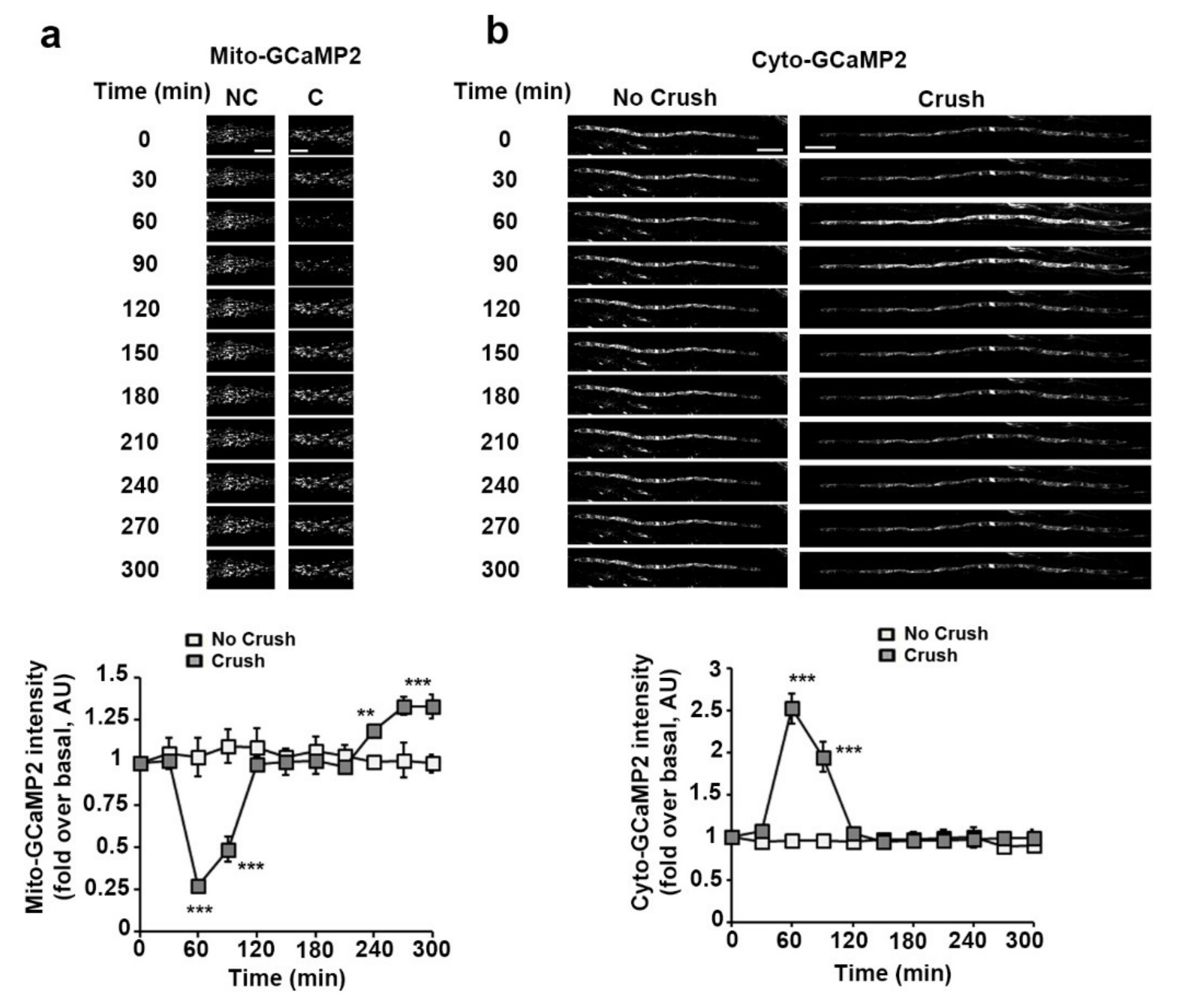
3.2. Sciatic Nerve Injury Induces Mitochondrial Calcium Release in the Cytoplasm, Mitochondria Slowdown and Matrix Alkalization in mSC
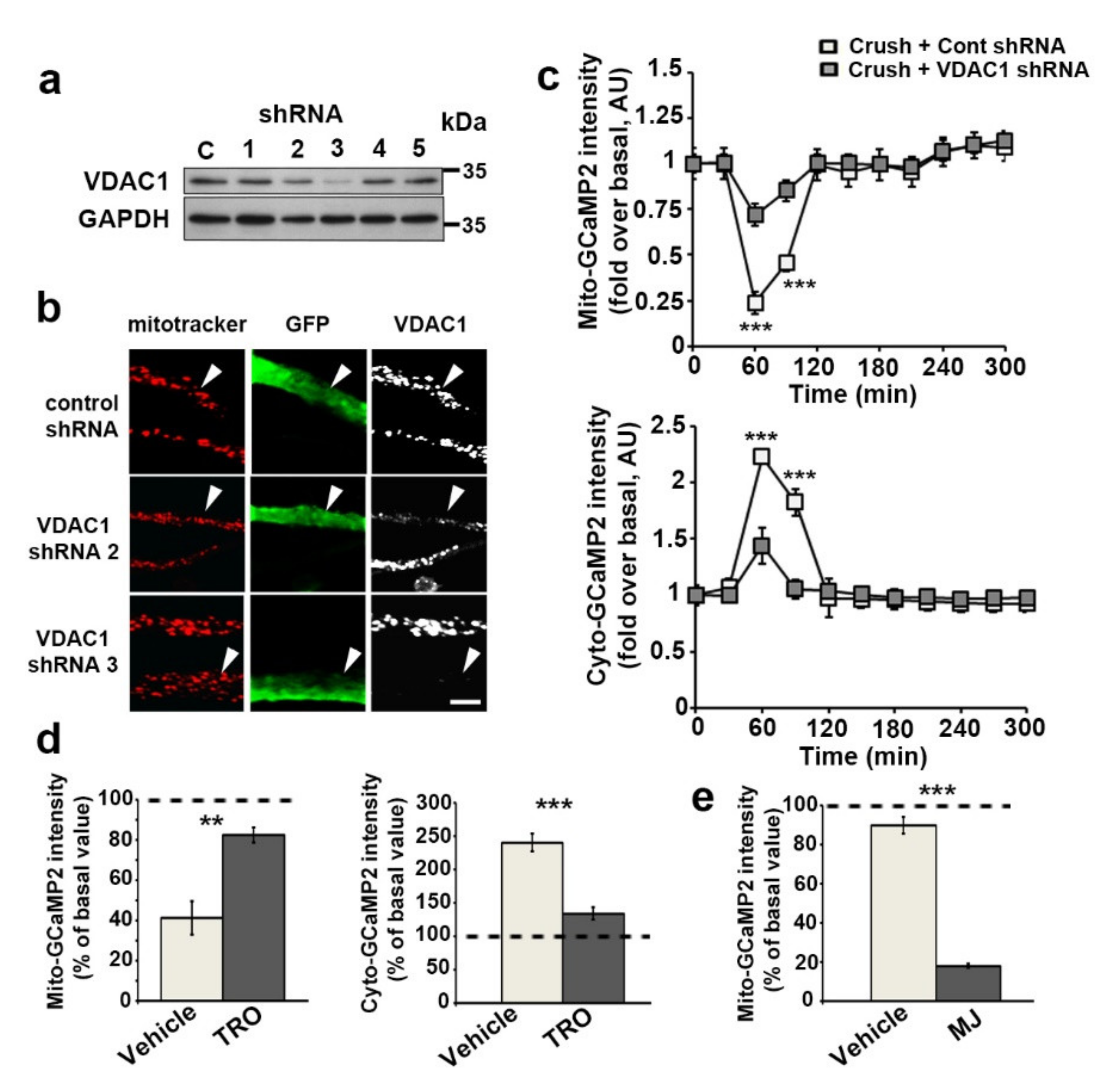
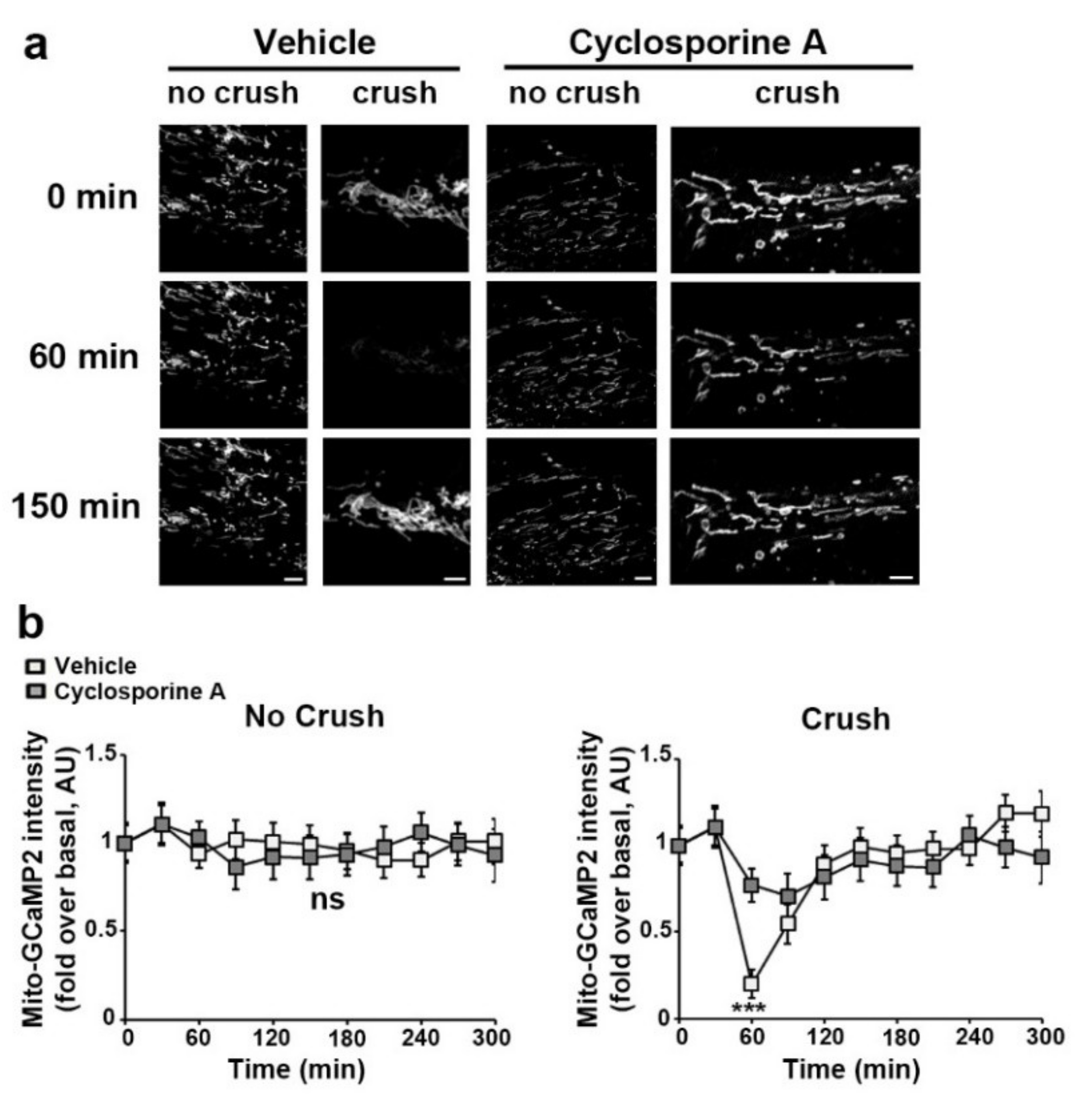
3.3. VDAC1 and mPTP Are Responsible for Mitochondrial Calcium Release in the Cytoplasm
3.4. The Release of Mitochondrial Calcium via VDAC1 Induces Schwann Cell Demyelination In Vivo via MAPK and CJUN ACTIVATION
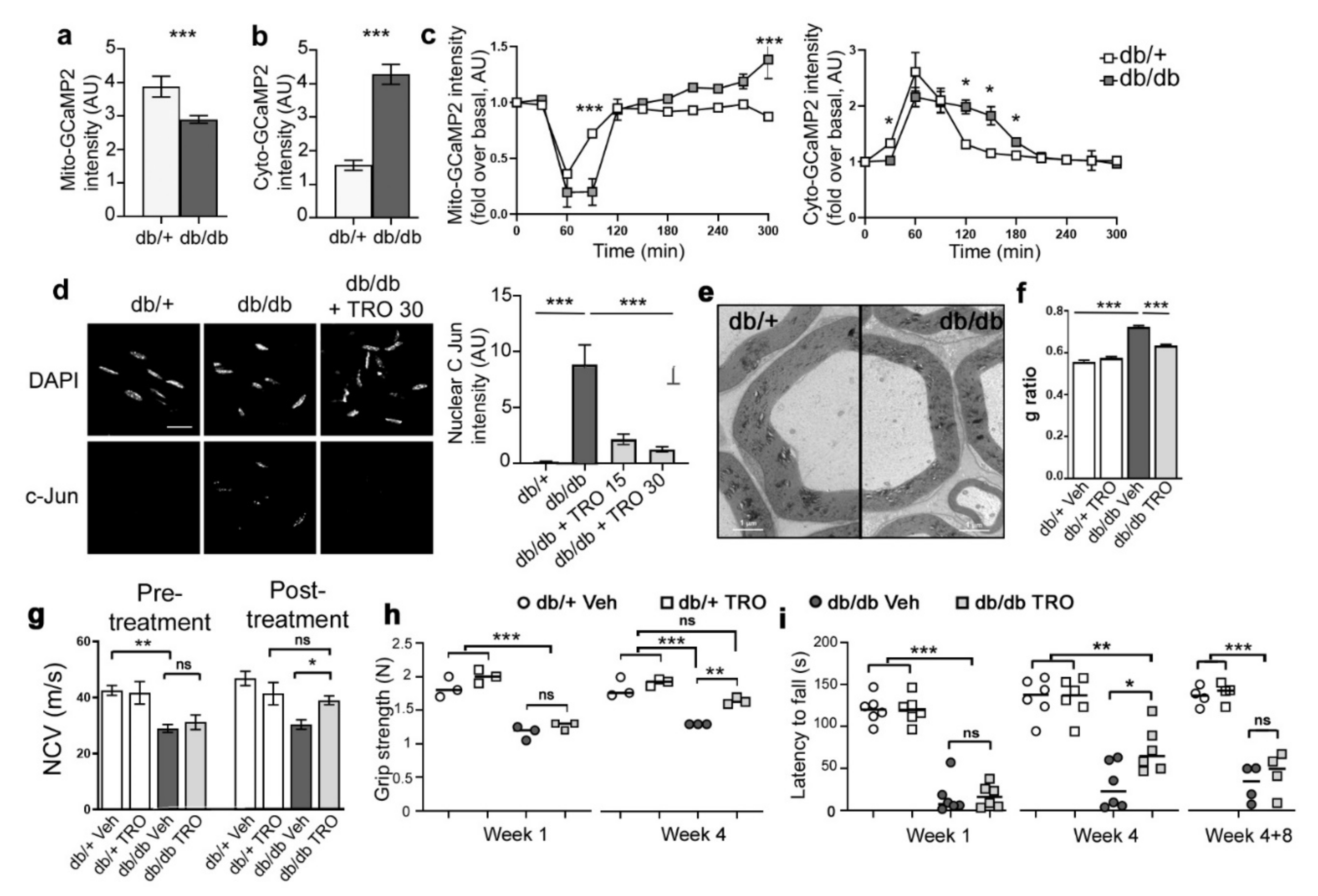
3.5. Basal Calcium Levels and Mitochondrial Calcium Release Kinetics Are Altered in db/db Diabetic Mice
3.6. Blocking Mitochondrial Calcium through VDAC Reduces Peripheral Nerve Defects and Peripheral Neuropathy in db/db Diabetic Mice
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nave, K.-A. Myelination and Support of Axonal Integrity by Glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.L.; Brophy, P.J. Mechanisms of Axon Ensheathment and Myelin Growth. Nat. Rev. Neurosci. 2005, 6, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Tricaud, N.; Park, H.T. Wallerian Demyelination: Chronicle of a Cellular Cataclysm. Cell. Mol. Life Sci. 2017, 74, 4049–4057. [Google Scholar] [CrossRef] [PubMed]
- Park, H.T.; Kim, J.K.; Tricaud, N. The Conceptual Introduction of the “Demyelinating Schwann Cell” in Peripheral Demyelinating Neuropathies. Glia 2019, 67, 571–581. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Repair Schwann Cell and Its Function in Regenerating Nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Hughes, R.A.C. Peripheral Neuropathy. BMJ Br. Med. J. 2002, 324, 466–469. [Google Scholar] [CrossRef]
- Zochodne, D.W. Diabetic Polyneuropathy: An Update. Curr. Opin. Neurol. 2008, 21, 527–533. [Google Scholar] [CrossRef]
- Pham, K.; Gupta, R. Understanding the Mechanisms of Entrapment Neuropathies. Review Article. Neurosurg. Focus 2009, 26, E7. [Google Scholar] [CrossRef]
- Skre, H. Genetic and Clinical Aspects of Charcot-Marie-Tooth’s Disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. C-Jun Reprograms Schwann Cells of Injured Nerves to Generate a Repair Cell Essential for Regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef]
- Della-Flora Nunes, G.; Wilson, E.R.; Marziali, L.N.; Hurley, E.; Silvestri, N.; He, B.; O’Malley, B.W.; Beirowski, B.; Poitelon, Y.; Wrabetz, L.; et al. Prohibitin 1 Is Essential to Preserve Mitochondria and Myelin Integrity in Schwann Cells. Nat. Commun. 2021, 12, 3285. [Google Scholar] [CrossRef]
- Della-Flora Nunes, G.; Wilson, E.R.; Hurley, E.; He, B.; O’Malley, B.W.; Poitelon, Y.; Wrabetz, L.; Feltri, M.L. Activation of MTORC1 and C-Jun by Prohibitin1 Loss in Schwann Cells May Link Mitochondrial Dysfunction to Demyelination. eLife 2021, 10, e66278. [Google Scholar] [CrossRef]
- Viader, A.; Golden, J.P.; Baloh, R.H.; Schmidt, R.E.; Hunter, D.A.; Milbrandt, J. Schwann Cell Mitochondrial Metabolism Supports Long-Term Axonal Survival and Peripheral Nerve Function. J. Neurosci. 2011, 31, 10128–10140. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic Oligodendrocytes Maintain Myelin and Long-Term Axonal Integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Sasaki, Y.; Hackett, A.R.; Kim, S.; Strickland, A.; Milbrandt, J. Dysregulation of NAD+ Metabolism Induces a Schwann Cell Dedifferentiation Program. J. Neurosci. 2018, 38, 6546–6562. [Google Scholar] [CrossRef]
- Kim, S.; Maynard, J.C.; Sasaki, Y.; Strickland, A.; Sherman, D.L.; Brophy, P.J.; Burlingame, A.L.; Milbrandt, J. Schwann Cell O-GlcNAc Glycosylation Is Required for Myelin Maintenance and Axon Integrity. J. Neurosci. 2016, 36, 9633–9646. [Google Scholar] [CrossRef]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, C.S.; Yang, K.; Gross, R.W.; Milbrandt, J. Aberrant Schwann Cell Lipid Metabolism Linked to Mitochondrial Deficits Leads to Axon Degeneration and Neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef]
- Kalman, B.; Lublin, F.D.; Alder, H. Impairment of Central and Peripheral Myelin in Mitochondrial Diseases. Mult. Scler. J. 1997, 2, 267–278. [Google Scholar] [CrossRef]
- Sarzi, E.; Angebault, C.; Seveno, M.; Gueguen, N.; Chaix, B.; Bielicki, G.; Boddaert, N.; Mausset-Bonnefont, A.-L.; Cazevieille, C.; Rigau, V.; et al. The Human OPA1delTTAG Mutation Induces Premature Age-Related Systemic Neurodegeneration in Mouse. Brain 2012, 135, 3599–3613. [Google Scholar] [CrossRef]
- Chao de la Barca, J.M.; Simard, G.; Sarzi, E.; Chaumette, T.; Rousseau, G.; Chupin, S.; Gadras, C.; Tessier, L.; Ferré, M.; Chevrollier, A.; et al. Targeted Metabolomics Reveals Early Dominant Optic Atrophy Signature in Optic Nerves of Opa1delTTAG/+ Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 812–820. [Google Scholar] [CrossRef]
- Cornblath, D.R.; Hoke, A. Recent Advances in HIV Neuropathy. Curr. Opin. Neurol. 2006, 19, 446–450. [Google Scholar] [CrossRef]
- Fernyhough, P.; Roy Chowdhury, S.K.; Schmidt, R.E. Mitochondrial Stress and the Pathogenesis of Diabetic Neuropathy. Expert Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef]
- He, T.-C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A Simplified System for Generating Recombinant Adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar] [CrossRef]
- Okada, T.; Nonaka-Sarukawa, M.; Uchibori, R.; Kinoshita, K.; Hayashita-Kinoh, H.; Nitahara-Kasahara, Y.; Takeda, S.; Ozawa, K. Scalable Purification of Adeno-Associated Virus Serotype 1 (AAV1) and AAV8 Vectors, Using Dual Ion-Exchange Adsorptive Membranes. Hum. Gene Ther. 2009, 20, 1013–1021. [Google Scholar] [CrossRef]
- Gonzalez, S.; Fernando, R.N.; Perrin-Tricaud, C.; Tricaud, N. In Vivo Introduction of Transgenes into Mouse Sciatic Nerve Cells in Situ Using Viral Vectors. Nat. Protoc. 2014, 9, 1160–1169. [Google Scholar] [CrossRef]
- Gonzalez, S.; Fernando, R.; Berthelot, J.; Perrin-Tricaud, C.; Sarzi, E.; Chrast, R.; Lenaers, G.; Tricaud, N. In Vivo Time-Lapse Imaging of Mitochondria in Healthy and Diseased Peripheral Myelin Sheath. Mitochondrion 2015, 23, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, H.; Boukhaddaoui, H.; Rizgui, A.; Sar, C.; Berthelot, J.; Perrin-Tricaud, C.; Rigneault, H.; Tricaud, N. Label-Free Non-Linear Microscopy to Measure Myelin Outcome in a Rodent Model of Charcot-Marie-Tooth Diseases. J. Biophotonics 2018, 11, e201800186. [Google Scholar] [CrossRef]
- Mytskaniuk, V.; Bardin, F.; Boukhaddaoui, H.; Rigneault, H.; Tricaud, N. Implementation of a Coherent Anti-Stokes Raman Scattering (CARS) System on a Ti:Sapphire and OPO Laser Based Standard Laser Scanning Microscope. J. Vis. Exp. 2016, e54262. [Google Scholar] [CrossRef] [PubMed]
- Ozçelik, M.; Cotter, L.; Jacob, C.; Pereira, J.A.; Relvas, J.B.; Suter, U.; Tricaud, N. Pals1 Is a Major Regulator of the Epithelial-like Polarization and the Extension of the Myelin Sheath in Peripheral Nerves. J. Neurosci. 2010, 30, 4120–4131. [Google Scholar] [CrossRef]
- Breckwoldt, M.O.; Armoundas, A.A.; Aon, M.A.; Bendszus, M.; O’Rourke, B.; Schwarzländer, M.; Dick, T.P.; Kurz, F.T. Mitochondrial Redox and PH Signaling Occurs in Axonal and Synaptic Organelle Clusters. Sci. Rep. 2016, 6, 23251. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, Y.; Hou, T.; Zhang, H.; Qu, A.; Wang, X. Differential Mitochondrial Calcium Responses in Different Cell Types Detected with a Mitochondrial Calcium Fluorescent Indicator, Mito-GCaMP2. Acta Biochim. Biophys. Sin. 2011, 43, 822–830. [Google Scholar] [CrossRef]
- Roma, L.P.; Duprez, J.; Takahashi, H.K.; Gilon, P.; Wiederkehr, A.; Jonas, J.-C. Dynamic Measurements of Mitochondrial Hydrogen Peroxide Concentration and Glutathione Redox State in Rat Pancreatic β-Cells Using Ratiometric Fluorescent Proteins: Confounding Effects of PH with HyPer but Not RoGFP1. Biochem. J. 2012, 441, 971–978. [Google Scholar] [CrossRef]
- Jacobs, J.M.; Cavanagh, J.B. Species Differences in Internode Formation Following Two Types of Peripheral Nerve Injury. J. Anat. 1969, 105, 295–306. [Google Scholar]
- Shoshan-Barmatz, V.; Gincel, D. The Voltage-Dependent Anion Channel. Cell Biochem. Biophys. 2003, 39, 279–292. [Google Scholar] [CrossRef]
- Ritchie, J.M. Voltage-Gated Cation and Anion Channels in Mammalian Schwann Cells and Astrocytes. J. Physiol. 1987, 82, 248–257. [Google Scholar]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the Crossroads of Cell Metabolism, Apoptosis and Cell Stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Bordet, T.; Berna, P.; Abitbol, J.-L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and Characterization of Cholest-4-En-3-One, Oxime (TRO19622), a Novel Drug Candidate for Amyotrophic Lateral Sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef]
- Rosa, J.C.; De Cerqueira César, J. Role of Hexokinase and VDAC in Neurological Disorders. Curr. Mol. Pharmacol. 2016, 9, 320–331. [Google Scholar] [CrossRef]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl Jasmonate Binds to and Detaches Mitochondria-Bound Hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is MPTP the Gatekeeper for Necrosis, Apoptosis, or Both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Boscolo, R.; Bindoli, A. Induction of Mitochondrial Permeability Transition by Auranofin, a Gold(I)-Phosphine Derivative. Br. J. Pharmacol. 2002, 136, 1162–1168. [Google Scholar] [CrossRef]
- Lee, H.J.; Shin, Y.K.; Park, H.T. Mitogen Activated Protein Kinase Family Proteins and C-Jun Signaling in Injury-Induced Schwann Cell Plasticity. Exp. Neurobiol. 2014, 23, 130–137. [Google Scholar] [CrossRef][Green Version]
- Napoli, I.; Noon, L.A.; Ribeiro, S.; Kerai, A.P.; Parrinello, S.; Rosenberg, L.H.; Collins, M.J.; Harrisingh, M.C.; White, I.J.; Woodhoo, A.; et al. A Central Role for the ERK-Signaling Pathway in Controlling Schwann Cell Plasticity and Peripheral Nerve Regeneration In Vivo. Neuron 2012, 73, 729–742. [Google Scholar] [CrossRef]
- Yang, D.P.; Kim, J.; Syed, N.; Tung, Y.; Bhaskaran, A.; Mindos, T.; Mirsky, R.; Jessen, K.R.; Maurel, P.; Parkinson, D.B.; et al. P38 MAPK Activation Promotes Denervated Schwann Cell Phenotype and Functions as a Negative Regulator of Schwann Cell Differentiation and Myelination. J. Neurosci. 2012, 32, 7158–7168. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A.; Woodhoo, A.; Lloyd, A.C.; Feltri, M.L.; Wrabetz, L.; Behrens, A.; Mirsky, R.; et al. C-Jun Is a Negative Regulator of Myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Cotter, L.; Ozçelik, M.; Jacob, C.; Pereira, J.A.; Locher, V.; Baumann, R.; Relvas, J.B.; Suter, U.; Tricaud, N. Dlg1-PTEN Interaction Regulates Myelin Thickness to Prevent Damaging Peripheral Nerve Overmyelination. Science 2010, 328, 1415–1418. [Google Scholar] [CrossRef]
- Bélanger, E.; Henry, F.P.; Vallée, R.; Randolph, M.A.; Kochevar, I.E.; Winograd, J.M.; Lin, C.P.; Côté, D. In Vivo Evaluation of Demyelination and Remyelination in a Nerve Crush Injury Model. Biomed. Opt. Express 2011, 2, 2698–2708. [Google Scholar] [CrossRef]
- Eftekharpour, E.; Fernyhough, P. Oxidative Stress and Mitochondrial Dysfunction Associated with Peripheral Neuropathy in Type 1 Diabetes. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Zenker, J.; Poirot, O.; de Preux Charles, A.-S.; Arnaud, E.; Médard, J.-J.; Lacroix, C.; Kuntzer, T.; Chrast, R. Altered Distribution of Juxtaparanodal Kv1.2 Subunits Mediates Peripheral Nerve Hyperexcitability in Type 2 Diabetes Mellitus. J. Neurosci. 2012, 32, 7493–7498. [Google Scholar] [CrossRef]
- Wang, L.; Chopp, M.; Szalad, A.; Liu, Z.; Bolz, M.; Alvarez, F.M.; Lu, M.; Zhang, L.; Cui, Y.; Zhang, R.L.; et al. Phosphodiesterase-5 Is a Therapeutic Target for Peripheral Neuropathy in Diabetic Mice. Neuroscience 2011, 193, 399–410. [Google Scholar] [CrossRef]
- Hamilton, R.T.; Bhattacharya, A.; Walsh, M.E.; Shi, Y.; Wei, R.; Zhang, Y.; Rodriguez, K.A.; Buffenstein, R.; Chaudhuri, A.R.; Van Remmen, H. Elevated Protein Carbonylation, and Misfolding in Sciatic Nerve from Db/Db and Sod1(-/-) Mice: Plausible Link between Oxidative Stress and Demyelination. PLoS ONE 2013, 8, e65725. [Google Scholar] [CrossRef]
- Woodhoo, A.; Alonso, M.B.D.; Droggiti, A.; Turmaine, M.; D’Antonio, M.; Parkinson, D.B.; Wilton, D.K.; Al-Shawi, R.; Simons, P.; Shen, J.; et al. Notch Controls Embryonic Schwann Cell Differentiation, Postnatal Myelination and Adult Plasticity. Nat. Neurosci. 2009, 12, 839–847. [Google Scholar] [CrossRef]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca(2+) Transfer from the ER to Mitochondria: When, How and Why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef]
- Chuderland, D.; Seger, R. Calcium Regulates ERK Signaling by Modulating Its Protein-Protein Interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef]
- Enslen, H.; Tokumitsu, H.; Stork, P.J.; Davis, R.J.; Soderling, T.R. Regulation of Mitogen-Activated Protein Kinases by a Calcium/Calmodulin-Dependent Protein Kinase Cascade. Proc. Natl. Acad. Sci. USA 1996, 93, 10803–10808. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between Mitogen-Activated Protein Kinases and Mitochondria in Cardiac Diseases: Therapeutic Perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef]
- Kulisz, A.; Chen, N.; Chandel, N.S.; Shao, Z.; Schumacker, P.T. Mitochondrial ROS Initiate Phosphorylation of P38 MAP Kinase during Hypoxia in Cardiomyocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1324–L1329. [Google Scholar] [CrossRef]
- Porter, G.A.; Makuck, R.F.; Rivkees, S.A. Reduction in Intracellular Calcium Levels Inhibits Myoblast Differentiation. J. Biol. Chem. 2002, 277, 28942–28947. [Google Scholar] [CrossRef] [PubMed]
- Hantke, J.; Chandler, D.; King, R.; Wanders, R.J.A.; Angelicheva, D.; Tournev, I.; McNamara, E.; Kwa, M.; Guergueltcheva, V.; Kaneva, R.; et al. A Mutation in an Alternative Untranslated Exon of Hexokinase 1 Associated with Hereditary Motor and Sensory Neuropathy—Russe (HMSNR). Eur. J. Hum. Genet. 2009, 17, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Weaver, D.; Hajnóczky, G. Control of Mitochondrial Motility and Distribution by the Calcium Signal: A Homeostatic Circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic Regulation of the Mitochondrial Proton Gradient during Cytosolic Calcium Elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Demaurex, N. Perspectives on: SGP Symposium on Mitochondrial Physiology and Medicine: The Renaissance of Mitochondrial PH. J. Gen. Physiol. 2012, 139, 415–423. [Google Scholar] [CrossRef]
- García-Sancho, J. The Coupling of Plasma Membrane Calcium Entry to Calcium Uptake by Endoplasmic Reticulum and Mitochondria. J. Physiol. 2014, 592, 261–268. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tricaud, N.; Gautier, B.; Berthelot, J.; Gonzalez, S.; Van Hameren, G. Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1. Biomedicines 2022, 10, 1447. https://doi.org/10.3390/biomedicines10061447
Tricaud N, Gautier B, Berthelot J, Gonzalez S, Van Hameren G. Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1. Biomedicines. 2022; 10(6):1447. https://doi.org/10.3390/biomedicines10061447
Chicago/Turabian StyleTricaud, Nicolas, Benoit Gautier, Jade Berthelot, Sergio Gonzalez, and Gerben Van Hameren. 2022. "Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1" Biomedicines 10, no. 6: 1447. https://doi.org/10.3390/biomedicines10061447
APA StyleTricaud, N., Gautier, B., Berthelot, J., Gonzalez, S., & Van Hameren, G. (2022). Traumatic and Diabetic Schwann Cell Demyelination Is Triggered by a Transient Mitochondrial Calcium Release through Voltage Dependent Anion Channel 1. Biomedicines, 10(6), 1447. https://doi.org/10.3390/biomedicines10061447