
Predicting High Blood Pressure Using DNA Methylome-Based Machine Learning Models



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. DNA Methylome Level Measurements
2.3. Nested Cross-Validation
2.4. DNA Methylome Data Preprocessing
2.5. Predictive Models
2.6. Model Evaluation
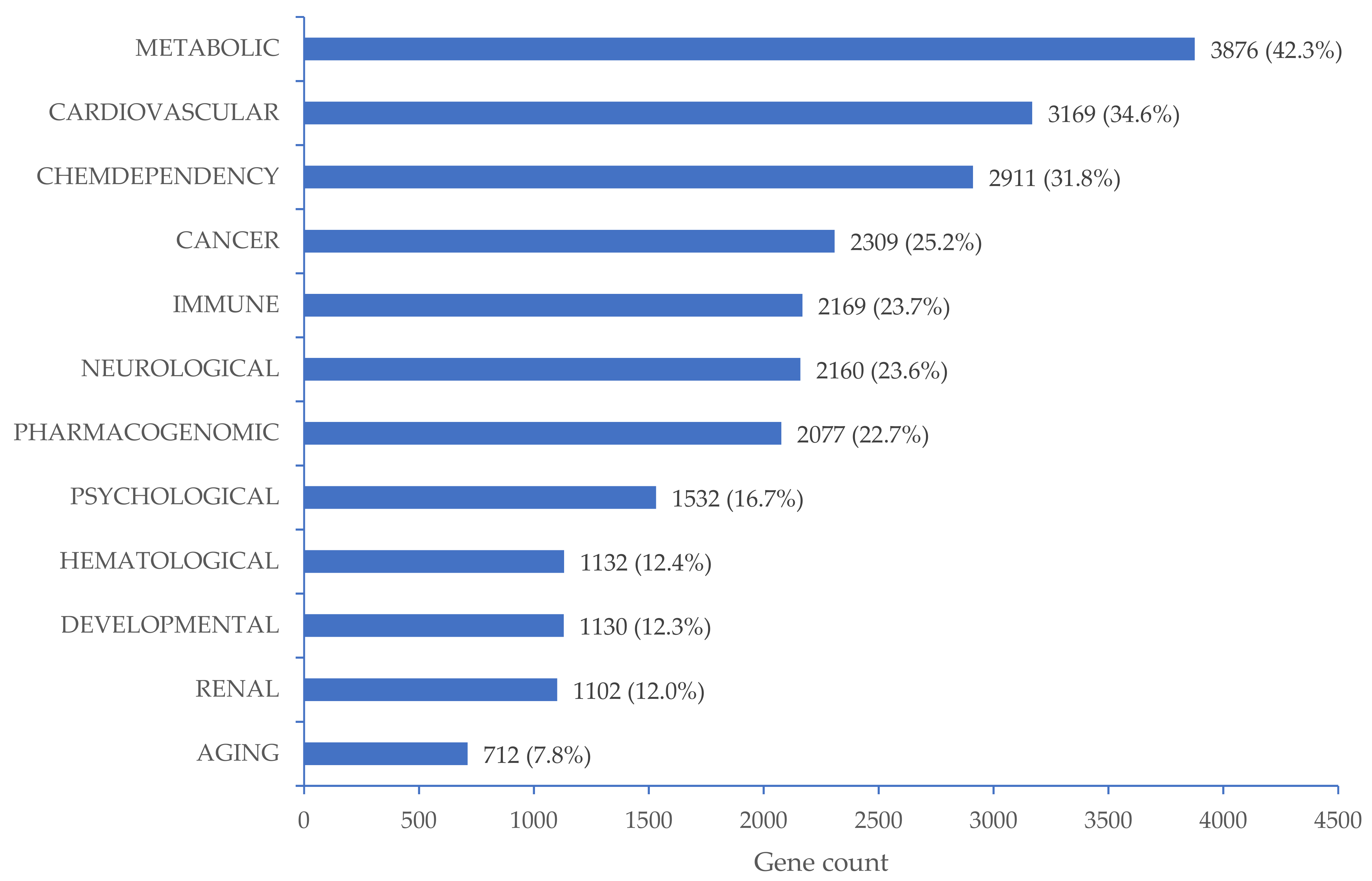
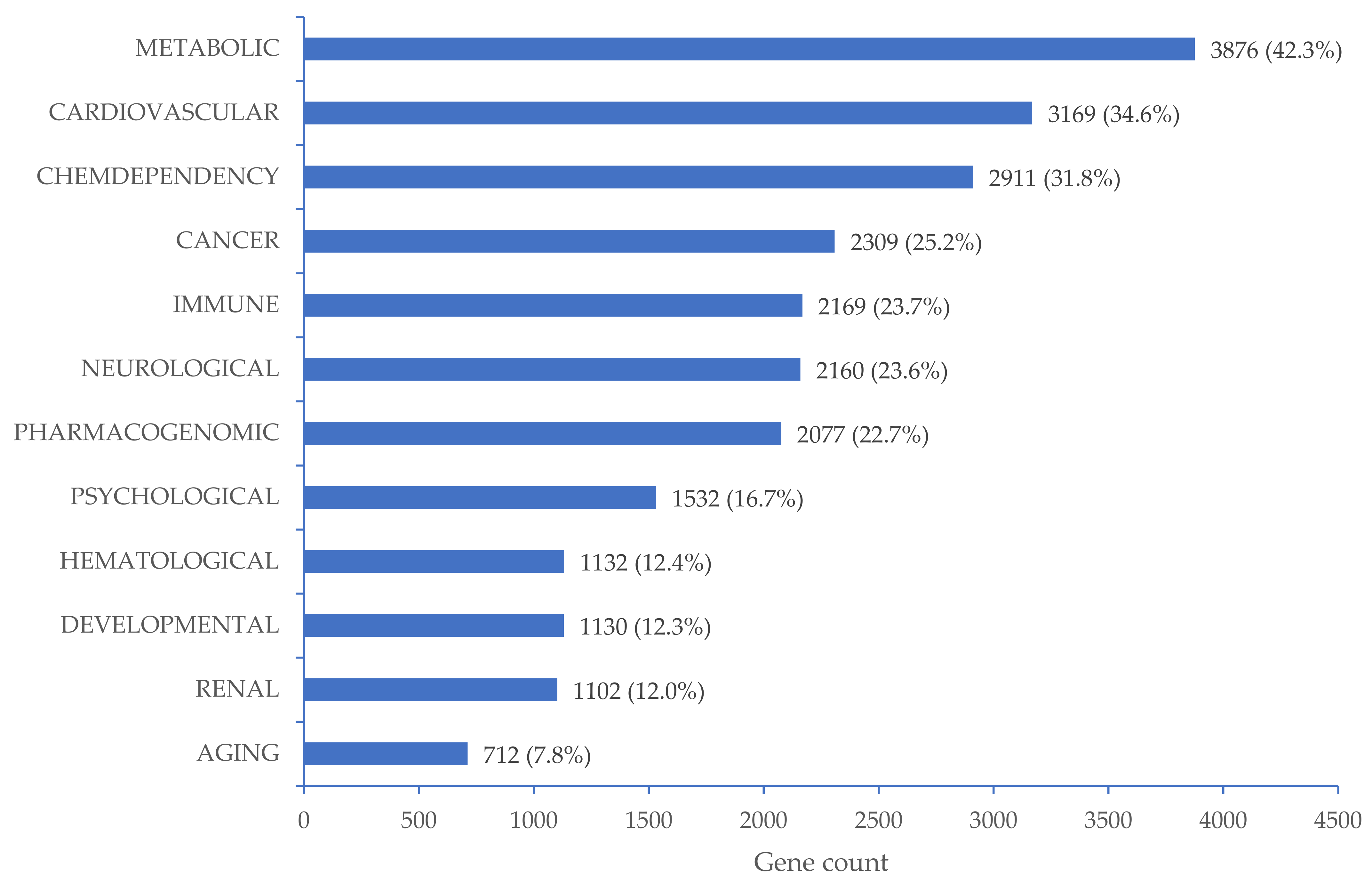
2.7. Functional Analyses
3. Results
3.1. Study Participants
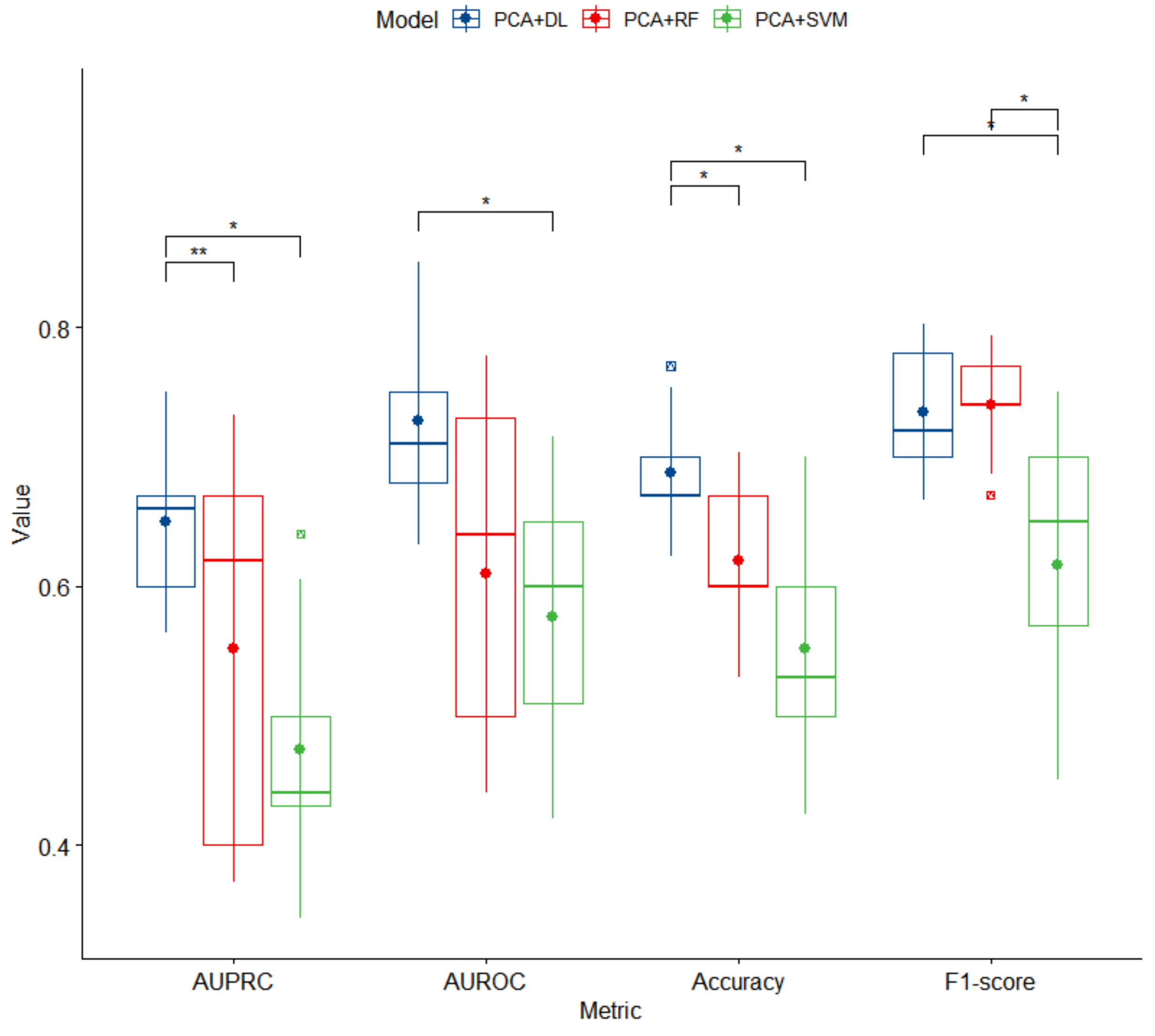
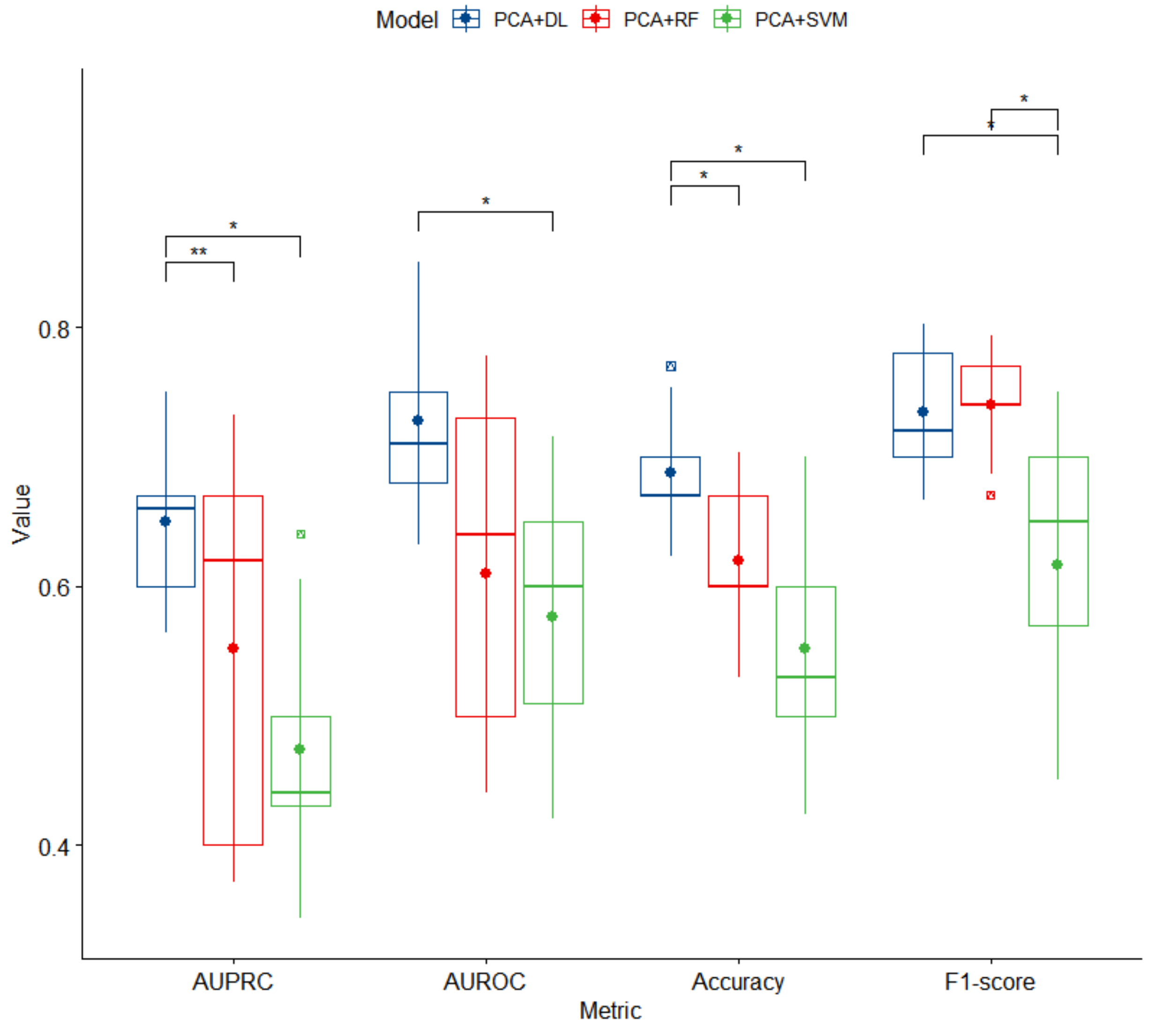
3.2. Predictive Models for High BP Detection
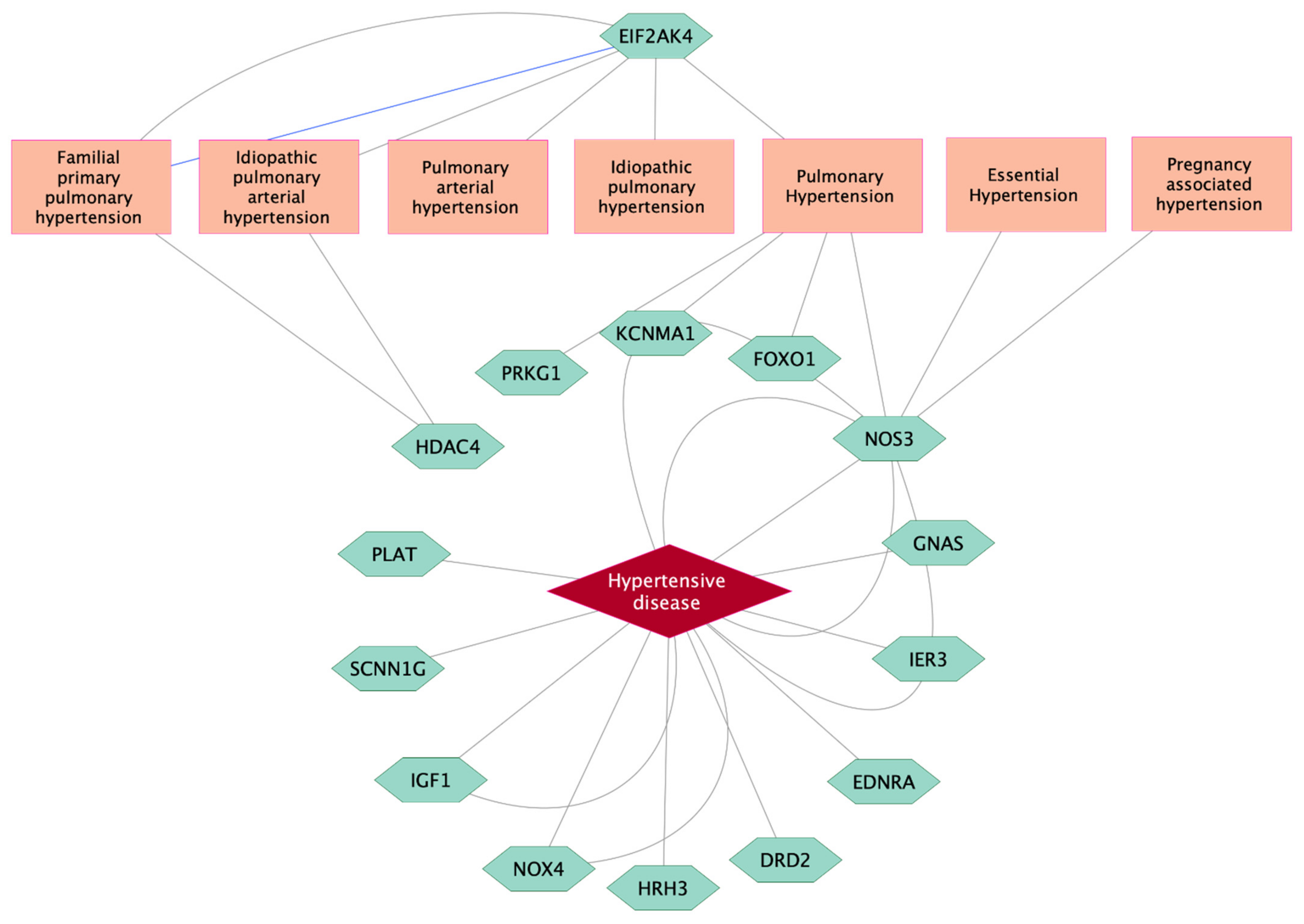
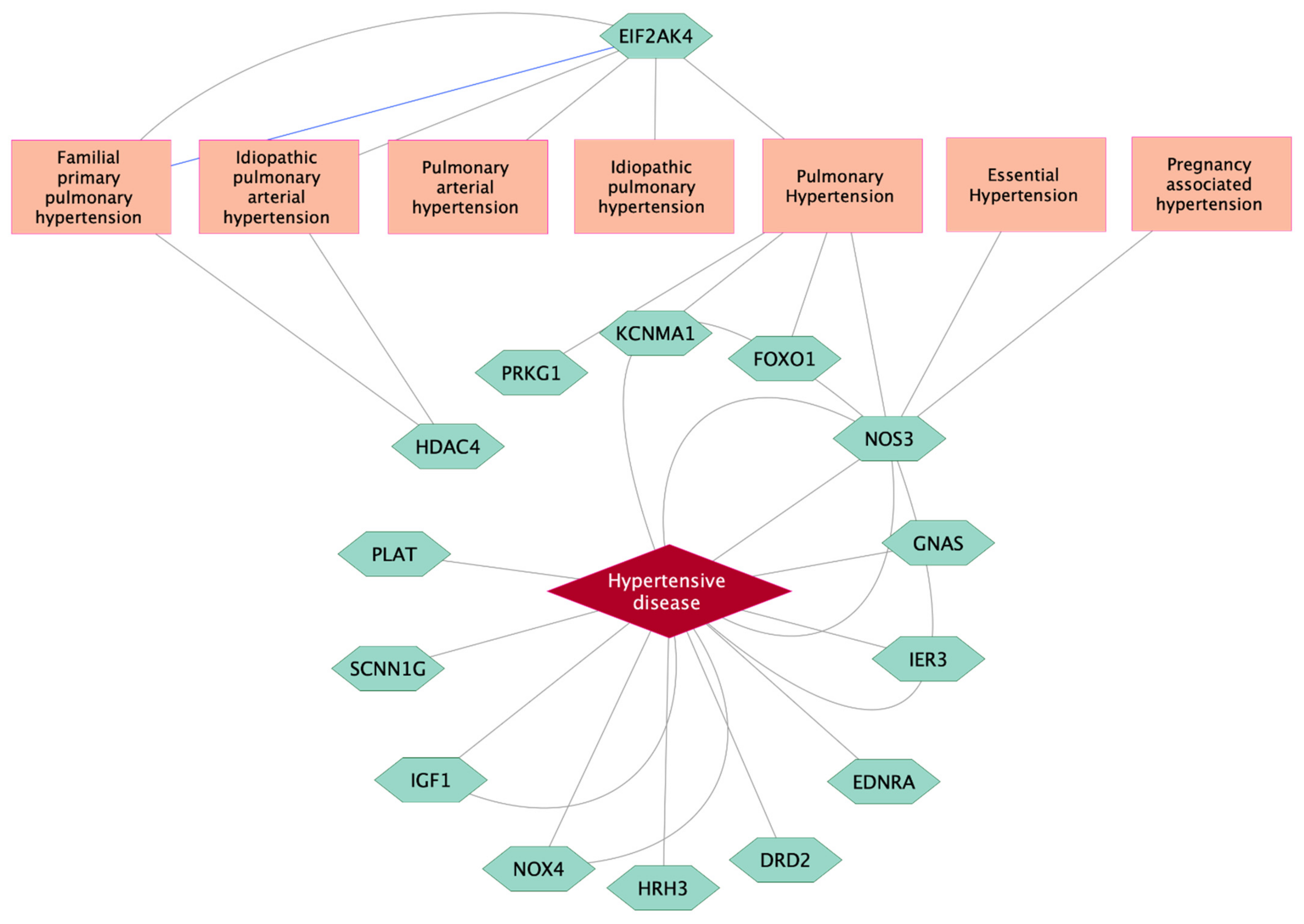
3.3. CpG Sites Significantly Associated with High BP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. A Global Brief on Hypertension: Silent Killer, Global Public Health Crisis: World Health Day 2013; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulec, S. Early diagnosis saves lives: Focus on patients with hypertension. Kidney Int. Suppl. 2013, 3, 332–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gong, L.; Tan, Y.; Hui, R.; Wang, Y. Hypertensive epigenetics: From DNA methylation to microRNAs. J. Hum. Hypertens. 2015, 29, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, Y.; Duan, S.; Perry, B.; Li, W.; He, Y. DNA methylation and hypertension: Emerging evidence and challenges. Brief Funct. Genomics 2016, 15, 460–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M. Epigenetic mechanisms and hypertension. Hypertension 2018, 72, 1244–1254. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. CpG Islands. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 205–207. [Google Scholar] [CrossRef]
- Richard, M.A.; Huan, T.; Ligthart, S.; Gondalia, R.; Jhun, M.A.; Brody, J.A.; Irvin, M.R.; Marioni, R.; Shen, J.; Tsai, P.C.; et al. DNA methylation analysis identifies loci for blood pressure regulation. Am. J. Hum. Genet. 2017, 101, 888–902. [Google Scholar] [CrossRef] [Green Version]
- Kato, N.; Loh, M.; Takeuchi, F.; Verweij, N.; Wang, X.; Zhang, W.; Kelly, T.N.; Saleheen, D.; Lehne, B.; Leach, I.M.; et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat. Genet. 2015, 47, 1282–1293. [Google Scholar] [CrossRef]
- Kazmi, N.; Elliott, H.R.; Burrows, K.; Tillin, T.; Hughes, A.D.; Chaturvedi, N.; Gaunt, T.R.; Relton, C.L. Associations between high blood pressure and DNA methylation. PLoS ONE 2020, 15, e0227728. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Yao, J.; Millstein, J.; Gao, L.; Siegmund, K.D.; Mack, W.; Whitfield-Maxwell, L.; Lurmann, F.; Hodis, H.; Avol, E.; et al. Prenatal air pollution exposures, DNA methyl transferase genotypes, and associations with newborn LINE1 and Alu methylation and childhood blood pressure and carotid intima-media thickness in the children’s health study. Environ. Health Perspect. 2016, 124, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Riviere, G.; Lienhard, D.; Andrieu, T.; Vieau, D.; Frey, B.M.; Frey, F.J. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics 2011, 6, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.N.; Liu, P.P.; Wang, L.; Yuan, F.; Xu, L.; Xin, Y.; Fei, L.J.; Zhong, Q.L.; Huang, Y.; Xu, L.; et al. Lower ADD1 gene promoter DNA methylation increases the risk of essential hypertension. PLoS ONE 2013, 8, e63455. [Google Scholar] [CrossRef]
- Fan, R.; Wang, W.J.; Zhong, Q.L.; Duan, S.W.; Xu, X.T.; Hao, L.M.; Zhao, J.; Zhang, L.N. Aberrant methylation of the GCK gene body is associated with the risk of essential hypertension. Mol. Med. Rep. 2015, 12, 2390–2394. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA Methylation as a biomarker for cardiovascular disease risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef] [Green Version]
- Kumar, I.; Singh, S.P. Chapter 26: Machine learning in bioinformatics. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 443–456. [Google Scholar]
- Casey, R.; Adelfio, A.; Connolly, M.; Wall, A.; Holyer, I.; Khaldi, N. Discovery through Machine Learning and Preclinical Validation of Novel Anti-Diabetic Peptides. Biomedicines 2021, 9, 276. [Google Scholar] [CrossRef]
- Tonkovic, P.; Kalajdziski, S.; Zdravevski, E.; Lameski, P.; Corizzo, R.; Pires, I.M.; Garcia, N.M.; Loncar-Turukalo, T.; Trajkovik, V. Literature on applied machine learning in metagenomic classification: A scoping review. Biology 2020, 9, 453. [Google Scholar] [CrossRef]
- Hsu, C.-W.; Tsai, S.-Y.; Wang, L.-J.; Liang, C.-S.; Carvalho, A.F.; Solmi, M.; Vieta, E.; Lin, P.-Y.; Hu, C.-A.; Kao, H.-Y. Predicting serum levels of lithium-treated patients: A supervised machine learning approach. Biomedicines 2021, 9, 1558. [Google Scholar] [CrossRef]
- Kaufmann, J.; Asalone, K.; Corizzo, R.; Saldanha, C.; Bracht, J.; Japkowicz, N. One-class ensembles for rare genomic sequences identification. In Proceedings of the 23rd International Conference on Discovery Science, Thessaloniki, Greece, 19–21 October 2020; Volume 12323, pp. 340–354. [Google Scholar] [CrossRef]
- Arslan, E.; Schulz, J.; Rai, K. Machine learning in epigenomics: Insights into cancer biology and medicine. Biochim. Biophys. Acta (BBA) 2021, 1876, 188588. [Google Scholar] [CrossRef]
- Brasil, S.; Neves, C.J.; Rijoff, T.; Falcao, M.; Valadao, G.; Videira, P.A.; Dos Reis Ferreira, V. Artificial intelligence in epigenetic studies: Shedding light on rare diseases. Front Mol. Biosci. 2021, 8, 648012. [Google Scholar] [CrossRef]
- Rauschert, S.; Raubenheimer, K.; Melton, P.E.; Huang, R.C. Machine learning and clinical epigenetics: A review of challenges for diagnosis and classification. Clin. Epigenetics 2020, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Holder, L.B.; Haque, M.M.; Skinner, M.K. Machine learning for epigenetics and future medical applications. Epigenetics 2017, 12, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Chen, Y.; Luo, C.; Meng, F. Machine learning methods in precision medicine targeting epigenetic diseases. Curr. Pharm. Des. 2018, 24, 3998–4006. [Google Scholar] [CrossRef]
- Iesato, A.; Nucera, C. Role of regulatory non-coding RNAs in aggressive thyroid cancer: Prospective applications of neural network analysis. Molecules 2021, 26, 3022. [Google Scholar] [CrossRef]
- Del Amor, R.; Colomer, A.; Monteagudo, C.; Naranjo, V. A deep embedded refined clustering approach for breast cancer distinction based on DNA methylation. Neural. Comput. Appl. 2021. [Google Scholar] [CrossRef]
- Si, Z.; Yu, H.; Ma, Z. Learning deep features for DNA methylation data analysis. IEEE Access 2016, 4, 2732–2737. [Google Scholar] [CrossRef]
- Duan, X.R.; Yang, Y.L.; Tan, S.J.; Wang, S.H.; Feng, X.L.; Cui, L.X.; Feng, F.F.; Yu, S.C.; Wang, W.; Wu, Y.J. Application of artificial neural network model combined with four biomarkers in auxiliary diagnosis of lung cancer. Med. Biol. Eng. Comput. 2017, 55, 1239–1248. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision cardiovascular medicine: Artificial intelligence and epigenetics for the pathogenesis and prediction of coarctation in neonates. J. Matern. Fetal Neonatal. Med. 2022, 35, 457–464. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Er, A.; Aydas, B.; Turkoglu, O.; Taskin, B.D.; Duman, M.; Yilmaz, D.; Radhakrishna, U. Artificial intelligence and the detection of pediatric concussion using epigenomic analysis. Brain Res. 2020, 1726, 146510. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, C.; Liu, H.; Zhang, Q.; Li, H. An attention-based deep learning method for schizophrenia patients classification using DNA methylation data. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2020, 2020, 172–175. [Google Scholar] [CrossRef]
- Bae, S.; Hong, Y.C. Exposure to bisphenol A from drinking canned beverages increases blood pressure: Randomized crossover trial. Hypertension 2015, 65, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Du, P.; Bilke, S.; Triche, T.; Bootwalla, M., Jr. Methylumi: Handle Illumina Methylation Data. R package version 2.39.0. 2021. Available online: https://bioconductor.riken.jp/packages/3.0/bioc/html/methylumi.html (accessed on 27 February 2022).
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeurwaerder, S.; Defrance, M.; Bizet, M.; Calonne, E.; Bontempi, G.; Fuks, F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2014, 15, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Narayan, S. The generalized sigmoid activation function: Competitive supervised learning. Inf. Sci. 1997, 99, 69–82. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef]
- Japkowicz, N.; Shah, M. Evaluating Learning Algorithms: A Classification Perspective; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- World Health Organization. Ageing and Health. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 27 February 2022).
- AlKaabi, L.A.; Ahmed, L.S.; Al Attiyah, M.F.; Abdel-Rahman, M.E. Predicting hypertension using machine learning: Findings from Qatar Biobank Study. PLoS ONE 2020, 15, e0240370. [Google Scholar] [CrossRef]
- Koshimizu, H.; Kojima, R.; Kario, K.; Okuno, Y. Prediction of blood pressure variability using deep neural networks. Int. J. Med. Inform. 2020, 136, 104067. [Google Scholar] [CrossRef]
- LaFreniere, D.; Zulkernine, F.; Barber, D.; Martin, K. Using machine learning to predict hypertension from a clinical dataset. In Proceedings of the 2016 IEEE Symposium Series on Computational Intelligence (SSCI), Athens, Greece, 6–9 December 2016; pp. 1–7. [Google Scholar]
- Ture, M.; Kurt, I.; Turhan Kurum, A.; Ozdamar, K. Comparing classification techniques for predicting essential hypertension. Expert. Syst. Appl. 2005, 29, 583–588. [Google Scholar] [CrossRef]
- López-Martínez, F.; Núñez-Valdez, E.R.; Crespo, R.G.; García-Díaz, V. An artificial neural network approach for predicting hypertension using NHANES data. Sci. Rep. 2020, 10, 10620. [Google Scholar] [CrossRef]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [Green Version]
- Gillberg, L.; Ling, C. The potential use of DNA methylation biomarkers to identify risk and progression of type 2 diabetes. Front. Endocrinol. 2015, 6, 43. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA methylation markers for pan–cancer prediction by deep learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Xiao, Y.; Wu, J.; Zhao, X.; Li, H. A convolutional neural network based ensemble method for cancer prediction using DNA methylation data. In Proceedings of the 2019 11th International Conference on Machine Learning and Computing 2019, Zhuhai, China, 22–24 February 2019; pp. 191–196. [Google Scholar]
- Soler-Botija, C.; Galvez-Monton, C.; Bayes-Genis, A. Epigenetic biomarkers in cardiovascular diseases. Front. Genet. 2019, 10, 950. [Google Scholar] [CrossRef]
- He, W.J.; Li, C.; Rao, D.C.; Hixson, J.E.; Huang, J.; Cao, J.; Rice, T.K.; Shimmin, L.C.; Gu, D.; Kelly, T.N. Associations of renin-angiotensin-aldosterone system genes with blood pressure changes and hypertension incidence. Am. J. Hypertens. 2015, 28, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Tsioufis, C.; Kordalis, A.; Flessas, D.; Anastasopoulos, I.; Tsiachris, D.; Papademetriou, V.; Stefanadis, C. Pathophysiology of resistant hypertension: The role of sympathetic nervous system. Int. J. Hypertens. 2011, 2011, 642416. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Vallon, V. Mineralocorticoid-induced sodium appetite and renal salt retention: Evidence for common signaling and effector mechanisms. Nephron. Physiol. 2014, 128, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Rangel, M.; dos Santos, J.C.; Ortiz, P.H.; Hirata, M.; Jasiulionis, M.G.; Araujo, R.C.; Ierardi, D.F.; Franco Mdo, C. Modification of epigenetic patterns in low birth weight children: Importance of hypomethylation of the ACE gene promoter. PLoS ONE 2014, 9, e106138. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Demura, M.; Cheng, Y.; Zhu, A.; Karashima, S.; Yoneda, T.; Demura, Y.; Maeda, Y.; Namiki, M.; Ono, K.; et al. Dynamic CCAAT/enhancer binding protein-associated changes of DNA methylation in the angiotensinogen gene. Hypertension 2014, 63, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.A.; Bennett, V. Adducin: A physical model with implications for function in assembly of spectrin-actin complexes. J. Biol. Chem. 1995, 270, 18990–18996. [Google Scholar] [CrossRef] [Green Version]
- Arlt, A.; Schafer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 2011, 90, 545–552. [Google Scholar] [CrossRef]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef]
- Zhao, Y.D.; Cai, L.; Mirza, M.K.; Huang, X.; Geenen, D.L.; Hofmann, F.; Yuan, J.X.J.; Zhao, Y.-Y. Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am. J. Pathol. 2012, 180, 2268–2275. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Zhang, K.; Wu, Y.; Xu, Z.; Yong, Q.C.; Kumar, R.; Baker, K.M.; Zhu, Q.; Chen, S.; Guo, S. Novel mechanism of blood pressure regulation by forkhead box class O1-mediated transcriptional control of hepatic angiotensinogen. Hypertension 2014, 64, 1131–1140. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Variables | Parameter (n = 50) |
|---|---|
| Age, year, mean ± SD | 72.5 ± 3.5 |
| Sex, n (%) | |
| Male | 3 (6.0) |
| Female | 47 (94.0) |
| BMI, kg/m2, n (%) | |
| 18.5–<23.0 | 11 (22.0) |
| 23.0–<25.0 | 14 (28.0) |
| 25.0–<30.0 | 22 (44.0) |
| 30.0–<35.0 | 3 (6.0) |
| Drinker, n (%) | |
| Yes | 12 (24.0) |
| No | 38 (76.0) |
| Smoker, n (%) | |
| Yes | 3 (6.0) |
| No | 47 (94.0) |
| History of hypertension, n (%) | |
| Yes | 26 (52.0) |
| No | 24 (48.0) |
| History of diabetes, n (%) | |
| Yes | 42 (84.0) |
| No | 8 (16.0) |
| Current SBP, mmHg, mean ± SD | 128.5 ± 66.5 |
| <130, n (%) | 71 (47.3) |
| 130–139, n (%) | 48 (32.0) |
| ≥140, n (%) | 31 (20.7) |
| Current DBP, mmHg, mean ± SD | 77.3 ± 38.5 |
| <85, n (%) | 125 (83.3) |
| 85–89, n (%) | 16 (10.7) |
| ≥90, n (%) | 9 (6.0) |
| Current high BP status, n (%) | |
| Yes | 87 (58.0) |
| No | 63 (42.0) |
| CpG | Chr | Position | USCS Gene | SBP | DBP | ||||
|---|---|---|---|---|---|---|---|---|---|
| Estimate | SE | p-Value | Estimate | SE | p-Value | ||||
| cg20203971 | 2 | 240171099 | HDAC4 | 443.9 | 101.0 | <0.001 | 205.5 | 56.0 | <0.001 |
| cg03573792 | 4 | 148465429 | EDNRA | 99.1 | 57.7 | 0.088 | 64.4 | 31.3 | 0.041 |
| cg04956913 | 6 | 30712436 | IER3 | –413.3 | 161.2 | 0.011 | –202.1 | 88.1 | 0.023 |
| cg13224213 | 7 | 150689881 | NOS3 | –86.3 | 41.7 | 0.040 | –57.7 | 22.5 | 0.012 |
| cg18899064 | 8 | 42066228 | PLAT | 58.2 | 70.4 | 0.410 | 91.3 | 37.6 | 0.016 |
| cg07528661 | 10 | 78647708 | KCNMA1 | 152.6 | 65.7 | 0.022 | 71.9 | 35.9 | 0.047 |
| cg06976598 | 10 | 53639124 | PRKG1 | –37.3 | 13.8 | 0.008 | –19.6 | 7.5 | 0.010 |
| cg18248586 | 11 | 113329026 | DRD2 | 64.4 | 33.4 | 0.056 | 47.2 | 18.0 | 0.010 |
| cg03793270 | 11 | 89224684 | NOX4 | 43.4 | 20.9 | 0.040 | 26.0 | 11.3 | 0.023 |
| cg16655193 | 12 | 102802953 | IGF1 | –115.9 | 52.5 | 0.029 | –52.5 | 28.7 | 0.070 |
| cg07109046 | 13 | 41204388 | FOXO1 | 50.8 | 15.1 | 0.001 | 23.1 | 8.3 | 0.006 |
| cg10821964 | 15 | 40269214 | EIF2AK4 | 177.0 | 70.2 | 0.013 | 102.4 | 38.1 | 0.008 |
| cg09094674 | 16 | 23194733 | SCNN1G | 78.7 | 30.3 | 0.010 | 64.0 | 16.0 | <0.001 |
| cg20019489 | 20 | 57414351 | GNAS | –47.0 | 16.6 | 0.005 | –21.1 | 9.1 | 0.022 |
| cg09640960 | 20 | 60794676 | HRH3 | 162.0 | 48.5 | 0.001 | 80.9 | 26.6 | 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.M.; Le, H.L.; Hwang, K.-B.; Hong, Y.-C.; Kim, J.H. Predicting High Blood Pressure Using DNA Methylome-Based Machine Learning Models. Biomedicines 2022, 10, 1406. https://doi.org/10.3390/biomedicines10061406
Nguyen TM, Le HL, Hwang K-B, Hong Y-C, Kim JH. Predicting High Blood Pressure Using DNA Methylome-Based Machine Learning Models. Biomedicines. 2022; 10(6):1406. https://doi.org/10.3390/biomedicines10061406
Chicago/Turabian StyleNguyen, Thi Mai, Hoang Long Le, Kyu-Baek Hwang, Yun-Chul Hong, and Jin Hee Kim. 2022. "Predicting High Blood Pressure Using DNA Methylome-Based Machine Learning Models" Biomedicines 10, no. 6: 1406. https://doi.org/10.3390/biomedicines10061406
APA StyleNguyen, T. M., Le, H. L., Hwang, K.-B., Hong, Y.-C., & Kim, J. H. (2022). Predicting High Blood Pressure Using DNA Methylome-Based Machine Learning Models. Biomedicines, 10(6), 1406. https://doi.org/10.3390/biomedicines10061406