
Mitochondrial Quality Control in the Heart: The Balance between Physiological and Pathological Stress



Abstract
:1. Energy Homeostasis
2. Mitochondrial Dynamics Maintain Energy Balance
3. Cardiac Mitochondria Respond to Energetic Stress
4. Physiological Adaptations
4.1. Acute Exercise
4.2. Chronic Exercise
5. Pathological Adaptations
5.1. Ischemic Heart Disease
5.2. Cardiac Hypertrophy and Pressure Overload-Induced Heart Failure
6. Dilated Cardiomyopathy
7. Pharmacological Modulation of Mitochondrial Dynamics and Quality Control
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harrell, C.; Gillespie, C.; Neigh, G. Energetic stress: The reciprocal relationship between energy availability and the stress response. Physiol. Behav. 2016, 166, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Bernstein, D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 2016, 60, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W., II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Von Stockum, S.; Marchesan, E.; Ziviani, E. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget 2018, 9, 12550–12551. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Moyzis, A.; Gustafsson, A.B. Multiple recycling routes: Canonical vs. non-canonical mitophagy in the heart. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 797–809. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906. [Google Scholar] [CrossRef]
- Yoo, B.; Lemaire, A.; Mangmool, S.; Wolf, M.J.; Curcio, A.; Mao, L.; Rockman, H.A. Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1377–H1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.L.; Fitzsimons, D.P. Frank-Starling relationship: Long on importance, short on mechanism. Circ. Res. 2002, 90, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, K.; Taberner, A.J.; Loiselle, D.S.; Han, J.C. Energetics equivalent of the cardiac force-length end-systolic zone: Implications for contractility and economy of contraction. Front. Physiol. 2019, 10, 1633. [Google Scholar] [CrossRef] [Green Version]
- Lövfors, W.; Ekström, J.; Jönsson, C.; Strålfors, P.; Cedersund, G.; Nyman, E. A systems biology analysis of lipolysis and fatty acid release from adipocytes in vitro and from adipose tissue in vivo. PLoS ONE 2021, 16, e0261681. [Google Scholar] [CrossRef]
- Mayer, S.E. Effect of catecholamines on cardiac metabolism. Circ. Res. 1974, 35 (Suppl. 3), 129–137. [Google Scholar] [CrossRef]
- Coronado, M.; Fajardo, G.; Nguyen, K.; Zhao, M.; Kooiker, K.; Jung, G.; Hu, D.-Q.; Reddy, S.; Sandoval, E.; Stotland, A.; et al. Physiological mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ. Res. 2018, 122, 282–295. [Google Scholar] [CrossRef]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Zhou, P.; Zhao, Y.T.; Guo, Y.B.; Xu, S.M.; Bai, S.H.; Lakatta, E.G.; Cheng, H.; Hao, X.-M.; Wang, S.-Q. Beta-adrenergic signaling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca2+ channel. Proc. Natl. Acad. Sci. USA 2009, 106, 18028–18033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortassa, S.; Aon, M.A.; Marban, E.; Winslow, R.L.; O’Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, S.; Xu, J.; Xin, Y.; Lu, Y.; Zhang, H.; Zhou, B.; Xu, H.; Sheu, S.-S.; Tian, R.; et al. Elevated MCU expression by CaMKIIdeltaB limits pathological cardiac remodeling. Circulation 2022, 145, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef]
- Ko, H.J.; Tsai, C.Y.; Chiou, S.J.; Lai, Y.L.; Wang, C.H.; Cheng, J.T.; Chuang, T.H.; Huang, C.F.; Kwan, A.L.; Loh, J.K.; et al. The phosphorylation status of Drp1-Ser637 by PKA in mitochondrial fission modulates mitophagy via PINK1/Parkin to exert multipolar spindles assembly during mitosis. Biomolecules 2021, 11, 424. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Fernandes, T.; Barauna, V.G.; Negrao, C.E.; Phillips, M.I.; Oliveira, E.M. Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H543–H552. [Google Scholar] [CrossRef] [Green Version]
- Rimbaud, S.; Garnier, A.; Ventura-Clapier, R. Mitochondrial biogenesis in cardiac pathophysiology. Pharmacol. Rep. 2009, 61, 131–138. [Google Scholar] [CrossRef]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.K.; Wang, Y.H.; Sun, L.; He, X.; Zhao, M.; Feng, Z.H.; Yu, X.J.; Zang, W.J. Aerobic interval training attenuates mitochondrial dysfunction in rats post-myocardial infarction: Roles of mitochondrial network dynamics. Int. J. Mol. Sci. 2014, 15, 5304–5322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Fiuza-Luces, C.; Delmiro, A.; Soares-Miranda, L.; Gonzalez-Murillo, A.; Martinez-Palacios, J.; Ramirez, M.; Lucia, A.; Moran, M. Exercise training can induce cardiac autophagy at end-stage chronic conditions: Insights from a graft-versus-host-disease mouse model. Brain Behav. Immun. 2014, 39, 56–60. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Schwalm, C.; Jamart, C.; Benoit, N.; Naslain, D.; Premont, C.; Prevet, J.; Van Thienen, R.; Deldicque, L.; Francaux, M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015, 29, 3515–3526. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, D.J. Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm. Sin. B 2020, 10, 1866–1879. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yan, W.; Zhao, X.; Jia, Q.; Wang, J.; Zhang, H.; Liu, C.; He, K.; Sun, Z. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell. Signal. 2019, 53, 1–13. [Google Scholar] [CrossRef]
- Liang, Q.; Kobayashi, S. Mitochondrial quality control in the diabetic heart. J. Mol. Cell. Cardiol. 2016, 95, 57–69. [Google Scholar] [CrossRef]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.; Cai, F.; Zhao, F.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef] [Green Version]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS ONE 2013, 8, e77713. [Google Scholar] [CrossRef] [Green Version]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, M.; Yan, R.; Shan, H.; Diao, J.; Wei, J. Inhibition of Drp1 attenuates mitochondrial damage and myocardial injury in Coxsackievirus B3 induced myocarditis. Biochem. Biophys. Res. Commun. 2017, 484, 550–556. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef]
- Luan, Y.; Feng, Q.; Chen, X.; Ren, K.D.; Yang, Y. Emerging role of mitophagy in the heart: Therapeutic potentials to modulate mitophagy in cardiac diseases. Oxidative Med. Cell. Longev. 2021, 2021, 3259963. [Google Scholar] [CrossRef]
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell. Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Turkieh, A.; El Masri, Y.; Pinet, F.; Dubois-Deruy, E. Mitophagy regulation following myocardial infarction. Cells 2022, 11, 199. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Zhou, H.; He, L.; Xu, G.; Chen, L. Mitophagy in cardiovascular disease. Clin. Chim. Acta 2020, 507, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.R.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS ONE 2013, 8, e62400. [Google Scholar]
- Xin, T.; Lu, C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging 2020, 12, 4474–4488. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Zhang, Y.; Zhao, Q.; Wang, X.; Lu, P.; Zhang, H.; Wang, Z.; Dong, H.; Zhang, Z. PEDF protects cardiomyocytes by promoting FUNDC1mediated mitophagy via PEDF-R under hypoxic condition. Int. J. Mol. Med. 2018, 41, 3394–3404. [Google Scholar] [PubMed] [Green Version]
- Feng, Y.; Madungwe, N.B.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef]
- Ji, W.; Wei, S.; Hao, P.; Xing, J.; Yuan, Q.; Wang, J.; Xu, F.; Chen, Y. Aldehyde dehydrogenase 2 has cardioprotective effects on myocardial ischaemia/reperfusion injury via suppressing mitophagy. Front. Pharmacol. 2016, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 Deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [Green Version]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin. Sci. 2019, 133, 497–513. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; Del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [PubMed] [Green Version]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. In Vitro 2018, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Makino, A.; Suarez, J.; Gawlowski, T.; Han, W.; Wang, H.; Scott, B.T.; Dillmann, W.H. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1296–R1302. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Li, J.; Shao, R.; Zhao, J.; Chen, M. FUNDC1: A Promising Mitophagy Regulator at the Mitochondria-Associated Membrane for Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 788634. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal. Res. 2019, 66, e12542. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nah, J.; Shirakabe, A.; Mukai, R.; Zhai, P.; Sung, E.A.; Ivessa, A.; Mizushima, W.; Nakada, Y.; Saito, T.; Hu, C.; et al. Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart. Cardiovasc. Res. 2022, 9, cvac003. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.; Lucena, J.R.; et al. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial morphology, dynamics, and function in human pressure overload or ischemic heart disease with preserved or reduced ejection fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Ashrafian, H.; Docherty, L.; Leo, V.; Towlson, C.; Neilan, M.; Steeples, V.; Lygate, C.A.; Hough, T.; Townsend, S.; Williams, D.; et al. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010, 6, e1001000. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Y.; Dorn, G.W., II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Vandeleur, D.; Chen, C.V.; Huang, E.J.; Connolly, A.J.; Sanchez, H.; Moon-Grady, A.J. Novel and lethal case of cardiac involvement in DNM1L mitochondrial encephalopathy. Am. J. Med. Genet. A 2019, 179, 2486–2489. [Google Scholar] [CrossRef]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burte, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell. Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef]
- Zhang, R.; Krigman, J.; Luo, H.; Ozgen, S.; Yang, M.; Sun, N. Mitophagy in cardiovascular homeostasis. Mech. Ageing Dev. 2020, 188, 111245. [Google Scholar] [CrossRef]
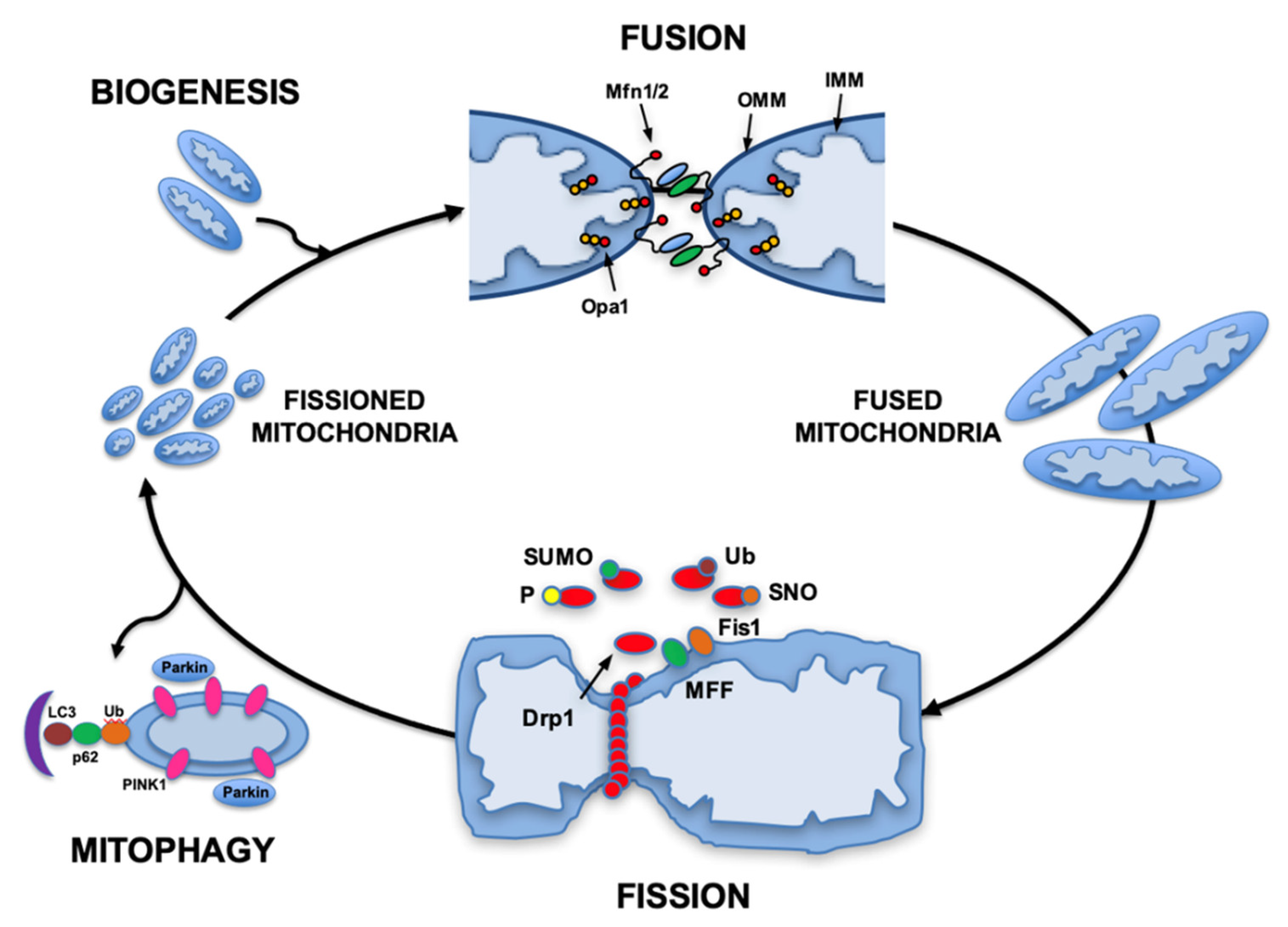

{kind=link}
| Disease | Mitochondrial Dynamics and Quality Control Alterations | Models | Phenotype |
|---|---|---|---|
| Ischemic Heart Disease |  Mitophagy Mitophagy | Parkin knockout | More sensitive to myocardial infarction [43] |
 Mitophagy Mitophagy | Parkin overexpression | Protected against hypoxia-mediated cell death [43] | |
| Mitophagy | PINK1 knockout | Larger myocardial infarcts than WT [62] | |
| Mitophagy | PINK1 overexpression | Reduced cell death after simulated IR [62] | |
| Mitophagy | Opa1 overexpression | Cardioprotective against hypoxia [63] | |
| Mitophagy | Pigment epithelial-derived factor | Cardioprotective effects hypoxia [64] | |
 Mitophagy Mitophagy | Post-ischemic G protein-coupled estrogen receptor 1 activation | Cardioprotective effects against IR [65] | |
 Mitophagy Mitophagy | ALDH2 activation with Alda-1 | Cardioprotective against IR [66] | |
| Mitophagy | Akap1 knockout | Larger infarct size, decreased survival [67] | |
| Fission | Neonatal murine cardiomyocytes and adult rat hearts after IR | Mitochondrial fragmentation and swelling within 30 min of IR [58,68] | |
| Fusion | Mitochondrial fusion promoter-M1 in IR | Cardioprotective against IR [69] | |
| Fusion | OPA1 overexpression | Protected from IR [70] | |
| Cardiac Hypertrophy and Failure | Mitophagy | Transverse aortic constriction | Mitophagy transiently activated at 3 to 7 days post transverse aortic constriction [71] |
| Mitophagy | Transverse aortic constriction | Mitophagy downregulated after 7 days post transverse aortic constriction [71] | |
| Fission | Stimulation of α1-adrenergic receptors with norepinephrine | Hypertrophy induced by norepinephrine with increased fission and decreased mitochondrial function [72] | |
| Fission | Dominant-negative Drp1 | Prevented fission and blocked norepinephrine hypertrophic growth [72] | |
| Mitophagy in heart failure | End-stage human heart failure | PINK1 protein levels are markedly reduced [73] | |
| Mitophagy in heart failure | Samples from heart failure patients | Isoform shift from AMPKα2 to AMPKα1 in failing heart, decreased mitophagy and mitochondrial function [74] | |
| Mitophagy in heart failure | AMPKα2 overexpression | Increase in cardiac mitophagy, improvement in mitochondrial function [74] | |
| Cardiomyopathies | Fission | DRP1 knockout/doxorubicin | Doxorubicin accelerates mitophagy flux, attenuated by DRP1 knockdown [47] |
| Fission | Isolated hearts/doxorubicin | Inhibition of mitochondrial fission with Mdivi-1 protects the heart against doxorubicin-induced cardiac injury [48] | |
| Mitophagy | Cells treated with doxorubicin | Doxorubicin induces mitophagy, activates the PINK1/Parkin pathway and inhibits the expression of PGC-1α [75] | |
| Mitophagy | High-fat diet induced diabetic cardiomyopathy | Activation of mitophagy protects against high fat induced diabetic cardiomyopathy [76] | |
| Fission | High glucose in neonatal cardiomyocytes | Decrease in mitochondrial membrane potential, overexpression of OPA1 attenuates mitochondrial fragmentation [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajardo, G.; Coronado, M.; Matthews, M.; Bernstein, D. Mitochondrial Quality Control in the Heart: The Balance between Physiological and Pathological Stress. Biomedicines 2022, 10, 1375. https://doi.org/10.3390/biomedicines10061375
Fajardo G, Coronado M, Matthews M, Bernstein D. Mitochondrial Quality Control in the Heart: The Balance between Physiological and Pathological Stress. Biomedicines. 2022; 10(6):1375. https://doi.org/10.3390/biomedicines10061375
Chicago/Turabian StyleFajardo, Giovanni, Michael Coronado, Melia Matthews, and Daniel Bernstein. 2022. "Mitochondrial Quality Control in the Heart: The Balance between Physiological and Pathological Stress" Biomedicines 10, no. 6: 1375. https://doi.org/10.3390/biomedicines10061375
APA StyleFajardo, G., Coronado, M., Matthews, M., & Bernstein, D. (2022). Mitochondrial Quality Control in the Heart: The Balance between Physiological and Pathological Stress. Biomedicines, 10(6), 1375. https://doi.org/10.3390/biomedicines10061375