
Chronic Rhinosinusitis, S. aureus Biofilm and Secreted Products, Inflammatory Responses, and Disease Severity

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Chronic Rhinosinusitis
1.2. Aetiology of CRS
1.3. CRS and Asthma
1.4. CRS and Dysbiosis
1.5. Staphylococcus aureus
1.6. Staphylococcal Biofilm
1.7. Staphylococcal Virulence Factors
1.7.1. Structural Factors
Adherence Factors (Adhesins)
Polysaccharide Capsule
1.7.2. Exoproteins
Pore-Forming and Enzymatic Toxins
Superantigens
1.8. Immune Response in CRS
1.8.1. Innate Immune Response
1.8.2. Adaptive Immune Response
1.9. CRS Inflammatory Endotypes
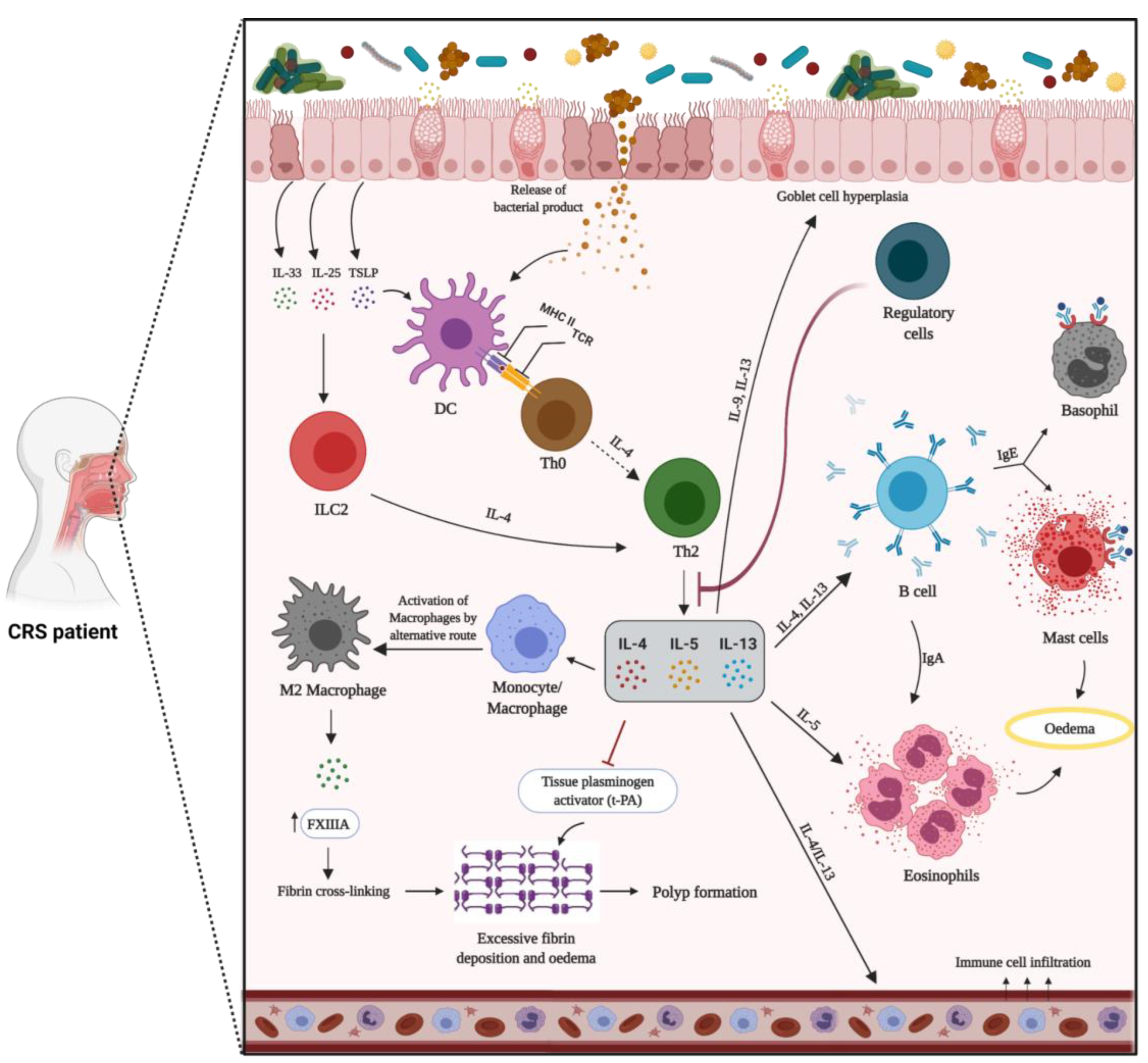
1.9.1. Mechanism of Type-2 Inflammation
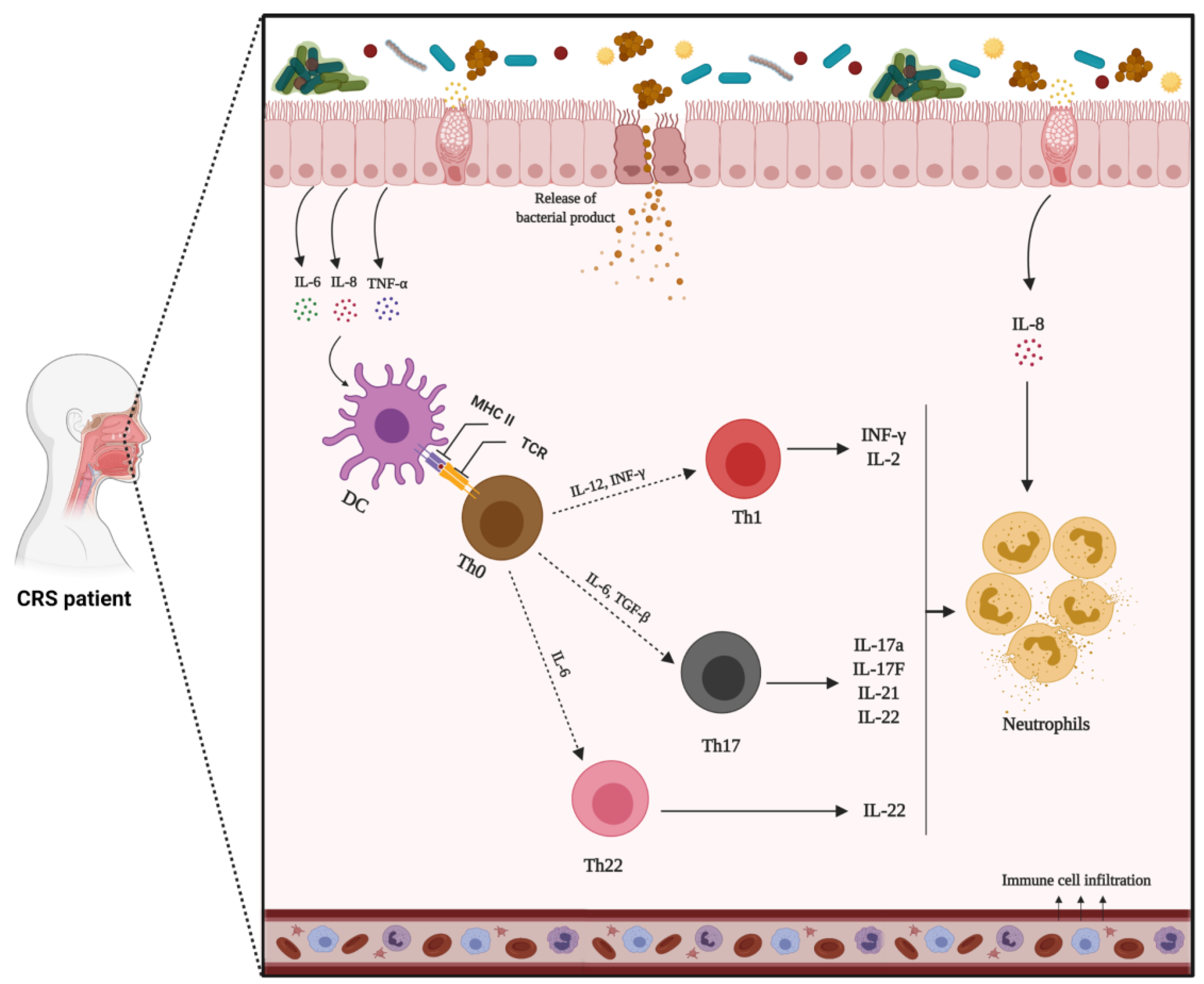
1.9.2. Mechanism of Non-Type 2 Inflammation
1.9.3. Mixed Inflammatory Patterns
1.10. Current Therapeutic Strategies for CRS
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fokkens, W.J.; Lund, V.J.; Mullol, J.; Bachert, C.; Alobid, I.; Baroody, F.; Cohen, N.; Cervin, A.; Douglas, R.; Gevaert, P. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology 2012, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.M.; Piccirillo, J.F.; Chandrasekhar, S.S.; Brook, I.; Ashok Kumar, K.; Kramper, M.; Orlandi, R.R.; Palmer, J.N.; Patel, Z.M.; Peters, A. Clinical practice guideline (update) adult sinusitis executive summary. Otolaryngol.-Head Neck Surg. 2015, 152, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Brook, I. Microbiology of chronic rhinosinusitis. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- DeConde, A.S.; Soler, Z.M. Chronic rhinosinusitis: Epidemiology and burden of disease. Am. J. Rhinol. Allergy 2016, 30, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lane, A.P. Chronic rhinosinusitis as a multifactorial inflammatory disorder. Curr. Infect. Dis. Rep. 2011, 13, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.W.; Schleimer, R.P.; Kern, R.C. Chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. Pract. 2016, 4, 565–572. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Ozdemir, C.; Akdis, M.; Akdis, C.A. Chronic rhinosinusitis: Pathogenesis, therapy options, and more. Expert Opin. Pharmacother. 2018, 19, 1805–1815. [Google Scholar] [CrossRef]
- Fastenberg, J.H.; Hsueh, W.D.; Mustafa, A.; Akbar, N.A.; Abuzeid, W.M. Biofilms in chronic rhinosinusitis: Pathophysiology and therapeutic strategies. World J. Otorhinolaryngol.-Head Neck Surg. 2016, 2, 219–229. [Google Scholar] [CrossRef]
- Chapurin, N.; Pynnonen, M.A.; Roberts, R.; Schulz, K.; Shin, J.J.; Witsell, D.L.; Parham, K.; Langman, A.; Carpenter, D.; Vambutas, A. CHEER national study of chronic rhinosinusitis practice patterns: Disease comorbidities and factors associated with surgery. Otolaryngol.–Head Neck Surg. 2017, 156, 751–756. [Google Scholar] [CrossRef]
- Frendø, M.; Håkansson, K.; Schwer, S.; Rix, I.; Ravn, A.T.; Backer, V.; von Buchwald, C. Asthma in ear, nose, and throat primary care patients with chronic rhinosinusitis with nasal polyps. Am. J. Rhinol. Allergy 2016, 30, e67–e71. [Google Scholar] [CrossRef]
- Benninger, M.S.; Sindwani, R.; Holy, C.E.; Hopkins, C. Impact of medically recalcitrant chronic rhinosinusitis on incidence of asthma. Proc. Int. Forum Allergy Rhinol. 2016, 6, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Chen, P.Y.; Lin, H.C.; Hung, S.H. Comorbidity profile of chronic rhinosinusitis: A population-based study. Laryngoscope 2014, 124, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, H.H.; Rotiroti, G. Allergic rhinitis, chronic rhinosinusitis and asthma: Unravelling a complex relationship. Curr. Opin. Otolaryngol. Head Neck Surg. 2013, 21, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, D.; Newson, R.; Lotvall, J.; Hastan, D.; Tomassen, P.; Keil, T.; Gjomarkaj, M.; Forsberg, B.; Gunnbjornsdottir, M.; Minov, J. Asthma in adults and its association with chronic rhinosinusitis: The GA2LEN survey in Europe. Allergy 2012, 67, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Kawata, R.; Haruna, S.; Moriyama, H.; Hirakawa, K.; Fujieda, S.; Masuyama, K.; Takenaka, H. Clinical epidemiological study of 553 patients with chronic rhinosinusitis in Japan. Allergol. Int. 2011, 60, 491–496. [Google Scholar] [CrossRef]
- Tomassen, P.; Vandeplas, G.; Van Zele, T.; Cardell, L.-O.; Arebro, J.; Olze, H.; Förster-Ruhrmann, U.; Kowalski, M.L.; Olszewska-Ziąber, A.; Holtappels, G. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J. Allergy Clin. Immunol. 2016, 137, 1449–1456.e4. [Google Scholar] [CrossRef]
- Langdon, C.; Mullol, J. Nasal polyps in patients with asthma: Prevalence, impact, and management challenges. J. Asthma Allergy 2016, 9, 45. [Google Scholar]
- Ryu, G.; Min, C.; Park, B.; Choi, H.G.; Mo, J.-H. Bidirectional association between asthma and chronic rhinosinusitis: Two longitudinal follow-up studies using a national sample cohort. Sci. Rep. 2020, 10, 9589. [Google Scholar] [CrossRef]
- Zhang, N.; Van Zele, T.; Perez-Novo, C.; Van Bruaene, N.; Holtappels, G.; DeRuyck, N.; Van Cauwenberge, P.; Bachert, C. Different types of T-effector cells orchestrate mucosal inflammation in chronic sinus disease. J. Allergy Clin. Immunol. 2008, 122, 961–968. [Google Scholar] [CrossRef]
- Klossek, J.M.; Neukirch, F.; Pribil, C.; Jankowski, R.; Serrano, E.; Chanal, I.; El Hasnaoui, A. Prevalence of nasal polyposis in France: A cross-sectional, case–control study. Allergy 2005, 60, 233–237. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Mullol, J.; Woessner, K.M.; Amin, N.; Mannent, L.P. Chronic rhinosinusitis with nasal polyps and asthma. J. Allergy Clin. Immunol. Pract. 2021, 9, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Zhang, N.; Holtappels, G.; De Lobel, L.; Van Cauwenberge, P.; Liu, S.; Lin, P.; Bousquet, J.; Van Steen, K. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J. Allergy Clin. Immunol. 2010, 126, 962–968.e966. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.R.; Feazel, L.M.; Gitomer, S.A.; Ir, D.; Robertson, C.E.; Frank, D.N. The microbiome of the middle meatus in healthy adults. PLoS ONE 2013, 8, e85507. [Google Scholar] [CrossRef] [PubMed]
- Bordin, A.; Sidjabat, H.E.; Cottrell, K.; Cervin, A. Chronic rhinosinusitis: A microbiome in dysbiosis and the search for alternative treatment options. Microbiol. Aust. 2016, 37, 149–152. [Google Scholar] [CrossRef]
- Feazel, L.M.; Robertson, C.E.; Ramakrishnan, V.R.; Frank, D.N. Microbiome complexity and Staphylococcus aureus in chronic rhinosinusitis. Laryngoscope 2012, 122, 467–472. [Google Scholar] [CrossRef]
- Abreu, N.A.; Nagalingam, N.A.; Song, Y.; Roediger, F.C.; Pletcher, S.D.; Goldberg, A.N.; Lynch, S.V. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci. Transl. Med. 2012, 4, 151ra124. [Google Scholar] [CrossRef]
- Choi, E.B.; Hong, S.W.; Kim, D.K.; Jeon, S.; Kim, K.R.; Cho, S.H.; Gho, Y.; Jee, Y.K.; Kim, Y.K. Decreased diversity of nasal microbiota and their secreted extracellular vesicles in patients with chronic rhinosinusitis based on a metagenomic analysis. Allergy 2014, 69, 517–526. [Google Scholar] [CrossRef]
- Ramakrishnan, V.R.; Feazel, L.M.; Abrass, L.J.; Frank, D.N. Prevalence and abundance of Staphylococcus aureus in the middle meatus of patients with chronic rhinosinusitis, nasal polyps, and asthma. Proc. Int. Forum Allergy Rhinol. 2013, 3, 267–271. [Google Scholar] [CrossRef]
- Ramakrishnan, V.R.; Hauser, L.J.; Feazel, L.M.; Ir, D.; Robertson, C.E.; Frank, D.N. Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. J. Allergy Clin. Immunol. 2015, 136, 334–342.e1. [Google Scholar] [CrossRef]
- Koutsourelakis, I.; Halderman, A.; Khalil, S.; Hittle, L.E.; Mongodin, E.F.; Lane, A.P. Temporal instability of the post-surgical maxillary sinus microbiota. BMC Infect. Dis. 2018, 18, 441. [Google Scholar] [CrossRef]
- Paramasivan, S.; Bassiouni, A.; Shiffer, A.; Dillon, M.R.; Cope, E.K.; Cooksley, C.; Ramezanpour, M.; Moraitis, S.; Ali, M.J.; Bleier, B. The international sinonasal microbiome study: A multicentre, multinational characterization of sinonasal bacterial ecology. Allergy 2020, 75, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Zhu, W.; Sartor, R.B.; Li, E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 2011, 19, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Vickery, T.W.; Ramakrishnan, V.R.; Suh, J.D. The role of Staphylococcus aureus in patients with chronic sinusitis and nasal polyposis. Curr. Allergy Asthma Rep. 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chalermwatanachai, T.; Zhang, N.; Holtappels, G.; Bachert, C. Association of mucosal organisms with patterns of inflammation in chronic rhinosinusitis. PLoS ONE 2015, 10, e0136068. [Google Scholar] [CrossRef] [PubMed]
- Van Zele, T.; Gevaert, P.; Watelet, J.-B.; Claeys, G.; Holtappels, G.; Claeys, C.; van Cauwenberge, P.; Bachert, C. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J. Allergy Clin. Immunol. 2004, 114, 981–983. [Google Scholar] [CrossRef]
- Schmidt, F.; Meyer, T.; Sundaramoorthy, N.; Michalik, S.; Surmann, K.; Depke, M.; Dhople, V.; Salazar, M.G.; Holtappels, G.; Zhang, N. Characterization of human and Staphylococcus aureus proteins in respiratory mucosa by in vivo-and immunoproteomics. J. Proteom. 2017, 155, 31–39. [Google Scholar] [CrossRef]
- Schwartz, J.S.; Peres, A.G.; Endam, L.M.; Cousineau, B.; Madrenas, J.; Desrosiers, M. Topical probiotics as a therapeutic alternative for chronic rhinosinusitis: A preclinical proof of concept. Am. J. Rhinol. Allergy 2016, 30, e202–e205. [Google Scholar] [CrossRef]
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323. [Google Scholar] [CrossRef]
- Seiberling, K.A.; Conley, D.B.; Tripathi, A.; Grammer, L.C.; Shuh, L.; Haines, G.K., III; Schleimer, R.; Kern, R.C. Superantigens and chronic rhinosinusitis: Detection of staphylococcal exotoxins in nasal polyps. Laryngoscope 2005, 115, 1580–1585. [Google Scholar] [CrossRef]
- Monaco, M.; de Araujo, F.P.; Cruciani, M.; Coccia, E.M.; Pantosti, A. Worldwide epidemiology and antibiotic resistance of Staphylococcus aureus. In Staphylococcus aureus; Springer: New York, NY, USA, 2016; pp. 21–56. [Google Scholar]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Drilling, A.; Coombs, G.W.; Tan, H.l.; Pearson, J.C.; Boase, S.; Psaltis, A.; Speck, P.; Vreugde, S.; Wormald, P.J. Cousins, siblings, or copies: The genomics of recurrent Staphylococcus aureus infections in chronic rhinosinusitis. Proc. Int. Forum Allergy Rhinol. 2014, 4, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Grumann, D.; Nübel, U.; Bröker, B.M. Staphylococcus aureus toxins–their functions and genetics. Infect. Genet. Evol. 2014, 21, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Teymournejad, O.; Montgomery, C.P. Evasion of immunological memory by S. aureus infection: Implications for vaccine design. Front. Immunol. 2021, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Neu, T.R.; Wozniak, D.J. The EPS matrix: The “house of biofilm cells”. J. Bacteriol. 2007, 189, 7945–7947. [Google Scholar] [CrossRef]
- Gil, C.; Solano, C.; Burgui, S.; Latasa, C.; García, B.; Toledo-Arana, A.; Lasa, I.; Valle, J. Biofilm matrix exoproteins induce a protective immune response against Staphylococcus aureus biofilm infection. Infect. Immun. 2014, 82, 1017–1029. [Google Scholar] [CrossRef]
- Singhal, D.; Psaltis, A.J.; Foreman, A.; Wormald, P.-J. The impact of biofilms on outcomes after endoscopic sinus surgery. Am. J. Rhinol. Allergy 2010, 24, 169–174. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Suh, J.D.; Ramakrishnan, V.; Palmer, J.N. Biofilms. Otolaryngol. Clin. N. Am. 2010, 43, 521–530. [Google Scholar] [CrossRef]
- Donné, J.; Dewilde, S. The challenging world of biofilm physiology. In Advances in Microbial Physiology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 67, pp. 235–292. [Google Scholar]
- Belas, R. Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 2014, 22, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M. Biofilms: Microbial life on surfaces. Emerg. Infect. Dis. 2002, 8, 881. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.D.; O’Toole, G.A. Second messenger regulation of biofilm formation: Breakthroughs in understanding c-di-GMP effector systems. Annu. Rev. Cell Dev. Biol. 2012, 28, 439–462. [Google Scholar] [CrossRef] [PubMed]
- Romeo, T. When the party is over: A signal for dispersal of Pseudomonas aeruginosa biofilms. J. Bacteriol. 2006, 188, 7325–7327. [Google Scholar] [CrossRef]
- Ramakrishnan, Y.; Shields, R.; Elbadawey, M.; Wilson, J. Biofilms in chronic rhinosinusitis: What is new and where next? J. Laryngol. Otol. 2015, 129, 744–751. [Google Scholar] [CrossRef]
- Singh, P.; Mehta, R.; Agarwal, S.; Mishra, P. Bacterial biofilm on the sinus mucosa of healthy subjects and patients with chronic rhinosinusitis (with or without nasal polyposis). J. Laryngol. Otol. 2015, 129, 46–49. [Google Scholar] [CrossRef]
- Sanderson, A.R.; Leid, J.G.; Hunsaker, D. Bacterial biofilms on the sinus mucosa of human subjects with chronic rhinosinusitis. Laryngoscope 2006, 116, 1121–1126. [Google Scholar] [CrossRef]
- Sanclement, J.A.; Webster, P.; Thomas, J.; Ramadan, H.H. Bacterial biofilms in surgical specimens of patients with chronic rhinosinusitis. Laryngoscope 2005, 115, 578–582. [Google Scholar] [CrossRef]
- Dlugaszewska, J.; Leszczynska, M.; Lenkowski, M.; Tatarska, A.; Pastusiak, T.; Szyfter, W. The pathophysiological role of bacterial biofilms in chronic sinusitis. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 1989–1994. [Google Scholar] [CrossRef]
- Prince, A.A.; Steiger, J.D.; Khalid, A.N.; Dogrhamji, L.; Reger, C.; Claire, S.E.; Chiu, A.G.; Kennedy, D.W.; Palmer, J.N.; Cohen, N.A. Prevalence of biofilm-forming bacteria in chronic rhinosinusitis. Am. J. Rhinol. 2008, 22, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Foreman, A.; Psaltis, A.J.; Tan, L.W.; Wormald, P.-J. Characterization of bacterial and fungal biofilms in chronic rhinosinusitis. Am. J. Rhinol. Allergy 2009, 23, 556–561. [Google Scholar] [CrossRef]
- Psaltis, A.J.; Weitzel, E.K.; Ha, K.R.; Wormald, P.-J. The effect of bacterial biofilms on post-sinus surgical outcomes. Am. J. Rhinol. 2008, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pratt, E.; Collins, A.M.; Sewell, W.A.; Harvey, R.J. Antigen selection in IgE antibodies from individuals with chronic rhinosinusitis with nasal polyps. Am. J. Rhinol. Allergy 2010, 24, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Cryer, J.; Schipor, I.; Perloff, J.R.; Palmer, J.N. Evidence of bacterial biofilms in human chronic sinusitis. J. Oto-Rhino-Laryngol. 2004, 66, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Claeys, S.E.; Tomassen, P.; Van Zele, T.; Zhang, N. Rhinosinusitis and asthma: A link for asthma severity. Curr. Allergy Asthma Rep. 2010, 10, 194–201. [Google Scholar] [CrossRef]
- Cirkovic, I.; Pavlovic, B.; Bozic, D.D.; Jotic, A.; Bakic, L.; Milovanovic, J. Antibiofilm effects of topical corticosteroids and intranasal saline in patients with chronic rhinosinusitis with nasal polyps depend on bacterial species and their biofilm-forming capacity. Eur. Arch. Oto-Rhino-Laryngol. 2017, 274, 1897–1903. [Google Scholar] [CrossRef]
- Mladina, R.; Skitarelić, N.; Musić, S.; Ristić, M. A biofilm exists on healthy mucosa of the paranasal sinuses: A prospectively performed, blinded, scanning electron microscope study. Clin. Otolaryngol. 2010, 35, 104–110. [Google Scholar] [CrossRef]
- Powers, M.E.; Wardenburg, J.B. Igniting the fire: Staphylococcus aureus virulence factors in the pathogenesis of sepsis. PLoS Pathog. 2014, 10, e1003871. [Google Scholar] [CrossRef]
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of virulence factors of Staphylococcus aureus: Novel function of known virulence factors that are implicated in activation of airway epithelial proinflammatory response. J. Pathog. 2011, 2011, 601905. [Google Scholar] [CrossRef]
- Foster, T.J. Surface proteins of Staphylococcus aureus. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Löffler, B.; Tuchscherr, L.; Niemann, S.; Peters, G. Staphylococcus aureus persistence in non-professional phagocytes. Int. J. Med. Microbiol. 2014, 304, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Moormeier, D.E.; Bayles, K.W. Staphylococcus aureus biofilm: A complex developmental organism. Mol. Microbiol. 2017, 104, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Morales, A.J.; Goodyear, C.S.; Silverman, G.J. Essential domain-dependent roles within soluble IgG for in vivo superantigen properties of staphylococcal protein A: Resolving the B-cell superantigen paradox. Front. Immunol. 2018, 9, 2011. [Google Scholar] [CrossRef] [PubMed]
- Guidry, A.; Oliver, S.; Squiggins, K.; Erbe, E.; Dowlen, H.; Hambleton, C.; Berning, L. Effect of anticapsular antibodies on neutrophil phagocytosis of Staphylococcus aureus. J. Dairy Sci. 1991, 74, 3360–3369. [Google Scholar] [CrossRef]
- Nanra, J.S.; Buitrago, S.M.; Crawford, S.; Ng, J.; Fink, P.S.; Hawkins, J.; Scully, I.L.; McNeil, L.K.; Aste-Amézaga, J.M.; Cooper, D. Capsular polysaccharides are an important immune evasion mechanism for Staphylococcus aureus. Hum. Vaccines Immunother. 2013, 9, 480–487. [Google Scholar] [CrossRef]
- Sutter, D.E.; Summers, A.M.; Keys, C.E.; Taylor, K.L.; Frasch, C.E.; Braun, L.E.; Fattom, A.I.; Bash, M.C. Capsular serotype of Staphylococcus aureus in the era of community-acquired MRSA. FEMS Immunol. Med. Microbiol. 2011, 63, 16–24. [Google Scholar] [CrossRef]
- Wilkinson, B.J.; Holmes, K.M. Staphylococcus aureus cell surface: Capsule as a barrier to bacteriophage adsorption. Infect. Immun. 1979, 23, 549–552. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Deghorain, M.; Van Melderen, L. The Staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef]
- Nepal, R.; Houtak, G.; Shaghayegh, G.; Bouras, G.; Shearwin, K.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Prophages encoding human immune evasion cluster genes are enriched in Staphylococcus aureus isolated from chronic rhinosinusitis patients with nasal polyps. Microb. Genom. 2021, 7, 000726. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.; Torres, V.J. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus α-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef]
- Caballero, A.R.; Foletti, D.L.; Bierdeman, M.A.; Tang, A.; Arana, A.M.; Hasa-Moreno, A.; Sangalang, E.R.B.; O’Callaghan, R.J. Effectiveness of alpha-toxin Fab monoclonal antibody therapy in limiting the pathology of Staphylococcus aureus keratitis. Ocul. Immunol. Inflamm. 2015, 23, 297–303. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef]
- Saftig, P.; Reiss, K. The “A Disintegrin and Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef]
- Wardenburg, J.B.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Jänicke, R.U. α-Toxin is a mediator of Staphylococcus aureus–induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 2001, 155, 637–648. [Google Scholar] [CrossRef]
- Poddighe, D.; Vangelista, L. Staphylococcus aureus infection and persistence in chronic rhinosinusitis: Focus on leukocidin ED. Toxins 2020, 12, 678. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus toxins and their molecular activity in infectious diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Yoong, P.; Torres, V.J. The effects of Staphylococcus aureus leukotoxins on the host: Cell lysis and beyond. Curr. Opin. Microbiol. 2013, 16, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Prevost, G.; Cribier, B.; Couppie, P.; Petiau, P.; Supersac, G.; Finck-Barbancon, V.; Monteil, H.; Piemont, Y. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect. Immun. 1995, 63, 4121–4129. [Google Scholar] [CrossRef]
- Morinaga, N.; Kaihou, Y.; Noda, M. Purification, cloning and characterization of variant LukE-LukD with strong leukocidal activity of staphylococcal bi-component leukotoxin family. Microbiol. Immunol. 2003, 47, 81–90. [Google Scholar] [CrossRef]
- Löffler, B.; Hussain, M.; Grundmeier, M.; Brück, M.; Holzinger, D.; Varga, G.; Roth, J.; Kahl, B.C.; Proctor, R.A.; Peters, G. Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 2010, 6, e1000715. [Google Scholar] [CrossRef]
- Alonzo, F., III; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef]
- Spaan, A.N.; van Strijp, J.A.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447. [Google Scholar] [CrossRef]
- DuMont, A.L.; Yoong, P.; Day, C.J.; Alonzo, F.; McDonald, W.H.; Jennings, M.P.; Torres, V.J. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. USA 2013, 110, 10794–10799. [Google Scholar] [CrossRef]
- Kaneko, J.; Kimura, T.; Kawakami, Y.; Tomita, T.; Kamio, Y. Panton-valentine leukocidin genes in a phage-like particle isolated from mitomycin C-treated Staphylococcus aureus V8 (ATCC 49775). Biosci. Biotechnol. Biochem. 1997, 61, 1960–1962. [Google Scholar] [CrossRef]
- Goerke, C.; Pantucek, R.; Holtfreter, S.; Schulte, B.; Zink, M.; Grumann, D.; Bröker, B.M.; Doskar, J.; Wolz, C. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 2009, 191, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Lina, G.; Piémont, Y.; Godail-Gamot, F.; Bes, M.; Peter, M.-O.; Gauduchon, V.; Vandenesch, F.; Etienne, J. Involvement of Panton-Valentine leukocidin—producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 1999, 29, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Gillet, Y.; Issartel, B.; Vanhems, P.; Fournet, J.-C.; Lina, G.; Bes, M.; Vandenesch, F.; Piémont, Y.; Brousse, N.; Floret, D. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 2002, 359, 753–759. [Google Scholar] [CrossRef]
- Shallcross, L.J.; Fragaszy, E.; Johnson, A.M.; Hayward, A.C. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 43–54. [Google Scholar] [CrossRef]
- Katayama, Y.; Baba, T.; Sekine, M.; Fukuda, M.; Hiramatsu, K. Beta-hemolysin promotes skin colonization by Staphylococcus aureus. J. Bacteriol. 2013, 195, 1194–1203. [Google Scholar] [CrossRef]
- Tajima, A.; Iwase, T.; Shinji, H.; Seki, K.; Mizunoe, Y. Inhibition of endothelial interleukin-8 production and neutrophil transmigration by Staphylococcus aureus beta-hemolysin. Infect. Immun. 2009, 77, 327–334. [Google Scholar] [CrossRef]
- Huseby, M.; Shi, K.; Brown, C.K.; Digre, J.; Mengistu, F.; Seo, K.S.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H.; Earhart, C.A. Structure and biological activities of beta toxin from Staphylococcus aureus. J. Bacteriol. 2007, 189, 8719–8726. [Google Scholar] [CrossRef]
- Walev, I.; Weller, U.; Strauch, S.; Foster, T.; Bhakdi, S. Selective killing of human monocytes and cytokine release provoked by sphingomyelinase (beta-toxin) of Staphylococcus aureus. Infect. Immun. 1996, 64, 2974–2979. [Google Scholar] [CrossRef]
- Huseby, M.J.; Kruse, A.C.; Digre, J.; Kohler, P.L.; Vocke, J.A.; Mann, E.E.; Bayles, K.W.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 14407–14412. [Google Scholar] [CrossRef]
- Otto, M. Phenol-soluble modulins. Int. J. Med. Microbiol. 2014, 304, 164–169. [Google Scholar] [CrossRef]
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Joo, H.-S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef] [PubMed]
- Baldry, M.; Bojer, M.S.; Najarzadeh, Z.; Vestergaard, M.; Meyer, R.L.; Otzen, D.E.; Ingmer, H. Phenol-soluble modulins modulate persister cell formation in Staphylococcus aureus. Front. Microbiol. 2020, 11, 573253. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Andreasen, M. Cross-talk between individual phenol-soluble modulins in Staphylococcus aureus biofilm enables rapid and efficient amyloid formation. Elife 2020, 9, e59776. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.-H.L.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 2007, 13, 1510–1514. [Google Scholar] [CrossRef]
- Ahmad-Mansour, N.; Loubet, P.; Pouget, C.; Dunyach-Remy, C.; Sotto, A.; Lavigne, J.-P.; Molle, V. Staphylococcus aureus toxins: An update on their pathogenic properties and potential treatments. Toxins 2021, 13, 677. [Google Scholar] [CrossRef]
- Mariutti, R.B.; Tartaglia, N.R.; Seyffert, N.; de Paula Castro, T.L.; Arni, R.K.; Azevedo, V.A.; Le Loir, Y.; Nishifuji, K. Exfoliative toxins of Staphylococcus aureus. Rise Virulence Antibiot. Resist. Staphylococcus Aureus 2017, 2, 1148–1165. [Google Scholar]
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef]
- Nishifuji, K.; Sugai, M.; Amagai, M. Staphylococcal exfoliative toxins:“Molecular scissors” of bacteria that attack the cutaneous defense barrier in mammals. J. Dermatol. Sci. 2008, 49, 21–31. [Google Scholar] [CrossRef]
- Morlock, B.; Spero, L.; Johnson, A. Mitogenic activity of staphylococcal exfoliative toxin. Infect. Immun. 1980, 30, 381–384. [Google Scholar] [CrossRef]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Van Zele, T.; Gevaert, P.; De Schrijver, L.; Van Cauwenberge, P. Superantigens and nasal polyps. Curr. Allergy Asthma Rep. 2003, 3, 523. [Google Scholar] [CrossRef] [PubMed]
- Heaton, T.; Mallon, D.; Venaille, T.; Holt, P. Staphylococcal enterotoxin induced IL-5 stimulation as a cofactor in the pathogenesis of atopic disease: The hygiene hypothesis in reverse? Allergy 2003, 58, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Van Zele, T.; Gevaert, P.; Holtappels, G.; Van Cauwenberge, P.; Bachert, C. Local immunoglobulin production in nasal polyposis is modulated by superantigens. Clin. Exp. Allergy 2007, 37, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Patou, J.; Gevaert, P.; Van Zele, T.; Holtappels, G.; Van Cauwenberge, P.; Bachert, C. Staphylococcus aureus enterotoxin B, protein A, and lipoteichoic acid stimulations in nasal polyps. J. Allergy Clin. Immunol. 2008, 121, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Zhang, N. Medical algorithm: Diagnosis and treatment of chronic rhinosinusitis. Allergy 2019, 75, 240–242. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, Y.; Wang, Z.C.; Ning, Q.; Liu, Z. Novel innate and adaptive lymphocytes: The new players in the pathogenesis of inflammatory upper airway diseases. Clin. Exp. Allergy 2018, 48, 620–631. [Google Scholar] [CrossRef]
- Stevens, W.W.; Lee, R.J.; Schleimer, R.P.; Cohen, N.A. Chronic rhinosinusitis pathogenesis. J. Allergy Clin. Immunol. 2015, 136, 1442–1453. [Google Scholar] [CrossRef]
- Hirschberg, A.; Kiss, M.; Kadocsa, E.; Polyanka, H.; Szabo, K.; Razga, Z.; Bella, Z.; Tiszlavicz, L.; Kemeny, L. Different activations of toll-like receptors and antimicrobial peptides in chronic rhinosinusitis with or without nasal polyposis. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 1779–1788. [Google Scholar] [CrossRef]
- Parker, D.; Prince, A. Innate immunity in the respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 189–201. [Google Scholar] [CrossRef]
- Soyka, M.B.; Wawrzyniak, P.; Eiwegger, T.; Holzmann, D.; Treis, A.; Wanke, K.; Kast, J.I.; Akdis, C.A. Defective epithelial barrier in chronic rhinosinusitis: The regulation of tight junctions by IFN-γ and IL-4. J. Allergy Clin. Immunol. 2012, 130, 1087–1096.e10. [Google Scholar] [CrossRef] [PubMed]
- Mahdavinia, M.; Keshavarzian, A.; Tobin, M.C.; Landay, A.; Schleimer, R.P. A comprehensive review of the nasal microbiome in chronic rhinosinusitis (CRS). Clin. Exp. Allergy 2016, 46, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556.e12. [Google Scholar] [CrossRef] [PubMed]
- Steelant, B.; Farré, R.; Wawrzyniak, P.; Belmans, J.; Dekimpe, E.; Vanheel, H.; Van Gerven, L.; Krohn, I.K.; Bullens, D.M.; Ceuppens, J.L. Impaired barrier function in patients with house dust mite–induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J. Allergy Clin. Immunol. 2016, 137, 1043–1053.e5. [Google Scholar] [CrossRef]
- El-Shazly, A.E.; Doloriert, H.C.; Bisig, B.; Lefebvre, P.; Delvenne, P.; Jacobs, N. Novel cooperation between CX 3 CL 1 and CCL 26 inducing NK cell chemotaxis via CX 3 CR 1: A possible mechanism for NK cell infiltration of the allergic nasal tissue. Clin. Exp. Allergy 2013, 43, 322–331. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, G.E.; Cho, G.S.; Kwon, H.-J.; Joo, C.H.; Kim, H.S.; Jang, Y.J. Natural killer cells from patients with chronic rhinosinusitis have impaired effector functions. PLoS ONE 2013, 8, e77177. [Google Scholar] [CrossRef]
- Awad, A.; Yassine, H.; Barrier, M.; Vorng, H.; Marquillies, P.; Tsicopoulos, A.; Duez, C. Natural killer cells induce eosinophil activation and apoptosis. PLoS ONE 2014, 9, e94492. [Google Scholar] [CrossRef]
- Thorén, F.B.; Riise, R.E.; Ousbäck, J.; Della Chiesa, M.; Alsterholm, M.; Marcenaro, E.; Pesce, S.; Prato, C.; Cantoni, C.; Bylund, J. Human NK Cells induce neutrophil apoptosis via an NKp46-and Fas-dependent mechanism. J. Immunol. 2012, 188, 1668–1674. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, Y.J. Role of natural killer cells in airway inflammation. Allergy Asthma Immunol. Res. 2018, 10, 448–456. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, G.E.; Lee, B.-J.; Kwon, S.W.; Lee, S.-H.; Kim, H.S.; Jang, Y.J. Natural killer cells regulate eosinophilic inflammation in chronic rhinosinusitis. Sci. Rep. 2016, 6, 27615. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Dyer, K.D.; Foster, P.S. Eosinophils: Changing perspectives in health and disease. Nat. Rev. Immunol. 2013, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Para, A.J.; Clayton, E.; Peters, A.T. Management of rhinosinusitis: An evidence based approach. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.C.; Weller, P.F. Contemporary understanding of the secretory granules in human eosinophils. J. Leukoc. Biol. 2018, 104, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.; Janson, C.; Bystrom, J. Role of eosinophil granulocytes in allergic airway inflammation endotypes. Scand. J. Immunol. 2016, 84, 75–85. [Google Scholar] [CrossRef]
- Frigas, E.; Motojima, S.; Gleich, G. The eosinophilic injury to the mucosa of the airways in the pathogenesis of bronchial asthma. Eur. Respir. J. Suppl. 1991, 13, 123s–135s. [Google Scholar]
- Tsuda, T.; Maeda, Y.; Nishide, M.; Koyama, S.; Hayama, Y.; Nojima, S.; Takamatsu, H.; Okuzaki, D.; Kinehara, Y.; Kato, Y. Eosinophil-derived neurotoxin enhances airway remodeling in eosinophilic chronic rhinosinusitis and correlates with disease severity. Int. Immunol. 2019, 31, 33–40. [Google Scholar] [CrossRef]
- Watelet, J.-B.; Bachert, C.; Claeys, C.; Van Cauwenberge, P. Matrix metalloproteinases MMP-7, MMP-9 and their tissue inhibitor TIMP-1: Expression in chronic sinusitis vs nasal polyposis. Allergy 2004, 59, 54–60. [Google Scholar] [CrossRef]
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.-U.; Bachert, C. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860.e6. [Google Scholar] [CrossRef]
- Hwang, C.S.; Park, S.C.; Cho, H.-J.; Park, D.-J.; Yoon, J.-H.; Kim, C.-H. Eosinophil extracellular trap formation is closely associated with disease severity in chronic rhinosinusitis regardless of nasal polyp status. Sci. Rep. 2019, 9, 8061. [Google Scholar] [CrossRef]
- Kountakis, S.E.; Arango, P.; Bradley, D.; Wade, Z.K.; Borish, L. Molecular and cellular staging for the severity of chronic rhinosinusitis. Laryngoscope 2004, 114, 1895–1905. [Google Scholar] [CrossRef]
- Vlaminck, S.; Vauterin, T.; Hellings, P.W.; Jorissen, M.; Acke, F.; Van Cauwenberge, P.; Bachert, C.; Gevaert, P. The importance of local eosinophilia in the surgical outcome of chronic rhinosinusitis: A 3-year prospective observational study. Am. J. Rhinol. Allergy 2014, 28, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Yoshikawa, M.; Asaka, D.; Okushi, T.; Matsuwaki, Y.; Otori, N.; Hema, T.; Moriyama, H. Mucosal eosinophilia and recurrence of nasal polyps—New classification of chronic rhinosinusitis. Rhinology 2011, 49, 392. [Google Scholar] [CrossRef] [PubMed]
- Mahdavinia, M.; Carter, R.G.; Ocampo, C.J.; Stevens, W.; Kato, A.; Tan, B.K.; Kern, R.C.; Conley, D.B.; Chandra, R.; Hulse, K.E. Basophils are elevated in nasal polyps of patients with chronic rhinosinusitis without aspirin sensitivity. J. Allergy Clin. Immunol. 2014, 133, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Kato, A.; Peters, A.T.; Suh, L.A.; Carter, R.; Norton, J.; Grammer, L.C.; Tan, B.K.; Chandra, R.K.; Conley, D.B. Glandular mast cells with distinct phenotype are highly elevated in chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2012, 130, 410–420.e5. [Google Scholar] [CrossRef]
- Zhai, G.T.; Li, J.X.; Zhang, X.H.; Liao, B.; Lu, X.; Liu, Z. Increased accumulation of CD30 ligand-positive mast cells associates with eosinophilic inflammation in nasal polyps. Laryngoscope 2019, 129, E110–E117. [Google Scholar] [CrossRef]
- Takabayashi, T.; Schleimer, R.P. Formation of nasal polyps: The roles of innate type 2 inflammation and deposition of fibrin. J. Allergy Clin. Immunol. 2020, 145, 740–750. [Google Scholar] [CrossRef]
- Belsky, M.A.; Corredera, E.; Banerjee, H.; Moore, J.; Wang, L.; Kane, L.P.; Lee, S.E. Association of mast cell burden and tim-3 expression with recalcitrant chronic rhinosinusitis with nasal polyps. Ann. Otol. Rhinol. Laryngol. 2021, 130, 1069–1077. [Google Scholar] [CrossRef]
- Corredera, E.; Phong, B.L.; Moore, J.A.; Kane, L.P.; Lee, S.E. TIM-3–Expressing Mast Cells Are Present in Chronic Rhinosinusitis with Nasal Polyps. Otolaryngol.–Head Neck Surg. 2018, 159, 581–586. [Google Scholar] [CrossRef]
- Hayes, S.M.; Howlin, R.; Johnston, D.A.; Webb, J.S.; Clarke, S.C.; Stoodley, P.; Harries, P.G.; Wilson, S.J.; Pender, S.L.; Faust, S.N. Intracellular residency of Staphylococcus aureus within mast cells in nasal polyps: A novel observation. J. Allergy Clin. Immunol. 2015, 135, 1648–1651.e5. [Google Scholar] [CrossRef]
- Gelardi, M.; Giancaspro, R.; Cassano, M.; Ribatti, D. The Underestimated Role of Mast Cells in the Pathogenesis of Rhinopathies. Int. Arch. Allergy Immunol. 2022, 183, 153–159. [Google Scholar] [CrossRef]
- Hayes, S.M.; Biggs, T.C.; Goldie, S.P.; Harries, P.G.; Walls, A.F.; Allan, R.N.; Pender, S.L.; Salib, R.J. Staphylococcus aureus internalization in mast cells in nasal polyps: Characterization of interactions and potential mechanisms. J. Allergy Clin. Immunol. 2020, 145, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Delemarre, T.; Bochner, B.S.; Simon, H.-U.; Bachert, C. Rethinking neutrophils and eosinophils in chronic rhinosinusitis. J. Allergy Clin. Immunol. 2021, 148, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.-Y.; Jiang, W.-X.; Liao, B.; Zhai, G.-T.; Wang, N.; Zhen, Z.; Ruan, J.-w.; Long, X.-B.; Wang, H. The activation and function of IL-36γ in neutrophilic inflammation in chronic rhinosinusitis. J. Allergy Clin. Immunol. 2018, 141, 1646–1658. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Teufelberger, A.R.; Nordengrün, M.; Braun, H.; Maes, T.; De Grove, K.; Holtappels, G.; O’Brien, C.; Provoost, S.; Hammad, H.; Gonçalves, A. The IL-33/ST2 axis is crucial in type 2 airway responses induced by Staphylococcus aureus–Derived serine protease–like protein D. J. Allergy Clin. Immunol. 2018, 141, 549–559.e7. [Google Scholar] [CrossRef]
- Kato, A. Immunopathology of chronic rhinosinusitis. Allergol. Int. 2015, 64, 121–130. [Google Scholar] [CrossRef]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Kao, S.S.T.; Ramezanpour, M.; Bassiouni, A.; Wormald, P.J.; Psaltis, A.J.; Vreugde, S. The effect of neutrophil serine proteases on human nasal epithelial cell barrier function. Int. Forum Allergy Rhinol. 2019, 9, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Fischer, B.M.; Malarkey, D.E.; Burch, L.H.; Wong, T.; Longphre, M.; Ho, S.B.; Foster, W.M. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L1293–L1302. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood J. Am. Soc. Hematol. 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, F.; Sun, Y.; Hong, H.; Wen, Y.; Lai, Y.; Xu, Z.; Luo, X.; Chen, Y.; Shi, J. LL-37 promotes neutrophil extracellular trap formation in chronic rhinosinusitis with nasal polyps. Clin. Exp. Allergy 2019, 49, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953. [Google Scholar] [CrossRef]
- Geissmann, F.; Gordon, S.; Hume, D.A.; Mowat, A.M.; Randolph, G.J. Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 2010, 10, 453. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 2011, 89, 557–563. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Holtappels, G.; Zhang, N.; Kubica, M.; Deswarte, K.; Derycke, L.; Claeys, S.; Hammad, H.; Brusselle, G.; Vandenabeele, P. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 2011, 66, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Poposki, J.A.; Uzzaman, A.; Nagarkar, D.R.; Chustz, R.T.; Peters, A.T.; Suh, L.A.; Carter, R.; Norton, J.; Harris, K.E.; Grammer, L.C. Increased expression of the chemokine CCL23 in eosinophilic chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2011, 128, 73–81.e4. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Kato, A.; Peters, A.T.; Hulse, K.E.; Suh, L.A.; Carter, R.; Norton, J.; Grammer, L.C.; Tan, B.K.; Chandra, R.K. Increased expression of factor XIII-A in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2013, 132, 584–592.e4. [Google Scholar] [CrossRef]
- Kortekaas Krohn, I.; Shikhagaie, M.M.; Golebski, K.; Bernink, J.; Breynaert, C.; Creyns, B.; Diamant, Z.; Fokkens, W.J.; Gevaert, P.; Hellings, P. Emerging roles of innate lymphoid cells in inflammatory diseases: Clinical implications. Allergy 2018, 73, 837–850. [Google Scholar] [CrossRef]
- Serafini, N.; Vosshenrich, C.A.; Di Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef]
- Mjösberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef]
- Vély, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef]
- Doherty, T.; Broide, D. Group 2 innate lymphoid cells: New players in human allergic diseases. J. Investig. Allergol. Clin. Immunol. 2015, 25, 1. [Google Scholar]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Bailey, M.; Zaunders, J.; Mrad, N.; Sacks, R.; Sewell, W.; Harvey, R. Group 2 innate lymphoid cells (ILC 2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin. Exp. Allergy 2015, 45, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; Van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25-and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Poposki, J.A.; Klingler, A.I.; Tan, B.K.; Soroosh, P.; Banie, H.; Lewis, G.; Hulse, K.E.; Stevens, W.W.; Peters, A.T.; Grammer, L.C. Group 2 innate lymphoid cells are elevated and activated in chronic rhinosinusitis with nasal polyps. Immun. Inflamm. Dis. 2017, 5, 233–243. [Google Scholar] [CrossRef]
- Walford, H.H.; Lund, S.J.; Baum, R.E.; White, A.A.; Bergeron, C.M.; Husseman, J.; Bethel, K.J.; Scott, D.R.; Khorram, N.; Miller, M. Increased ILC2s in the eosinophilic nasal polyp endotype are associated with corticosteroid responsiveness. Clin. Immunol. 2014, 155, 126–135. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef]
- Wherry, E.J.; Masopust, D. Adaptive Immunity: Neutralizing, Eliminating, and Remembering for the Next Time. In Viral Pathogenesis; Elsevier: Amsterdam, The Netherlands, 2016; pp. 57–69. [Google Scholar]
- Wegrzyn, A.S.; Jakiela, B.; Rückert, B.; Jutel, M.; Akdis, M.; Sanak, M.; Akdis, C.A. T-cell regulation during viral and nonviral asthma exacerbations. J. Allergy Clin. Immunol. 2015, 136, 194–197.e9. [Google Scholar] [CrossRef]
- Van Zele, T.; Claeys, S.; Gevaert, P.; Van Maele, G.; Holtappels, G.; Van Cauwenberge, P.; Bachert, C. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy 2006, 61, 1280–1289. [Google Scholar] [CrossRef]
- Roongrotwattanasiri, K.; Pawankar, R.; Kimura, S.; Mori, S.; Nonaka, M.; Yagi, T. Decreased expression of FOXP3 in nasal polyposis. Allergy Asthma Immunol. Res. 2012, 4, 24–30. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.M.; Munoz, A.; Hwang, P.H.; Nadeau, K.C. Migration of regulatory T cells toward airway epithelial cells is impaired in chronic rhinosinusitis with nasal polyposis. Clin. Immunol. 2010, 137, 111–121. [Google Scholar] [CrossRef]
- Van Bruaene, N.; Pérez-Novo, C.A.; Basinski, T.M.; Van Zele, T.; Holtappels, G.; De Ruyck, N.; Schmidt-Weber, C.; Akdis, C.; Van Cauwenberge, P.; Bachert, C. T-cell regulation in chronic paranasal sinus disease. J. Allergy Clin. Immunol. 2008, 121, 1435–1441.e3. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Zhang, N.; Zhang, J.; Krysko, O.; Zhang, Q.; Xian, J.; Derycke, L.; Qi, Y.; Li, K.; Liu, S. Forkhead box protein 3 in human nasal polyp regulatory T cells is regulated by the protein suppressor of cytokine signaling 3. J. Allergy Clin. Immunol. 2013, 132, 1314–1321.e3. [Google Scholar] [CrossRef] [PubMed]
- Tantilipikorn, P.; Sookrung, N.; Muangsomboon, S.; Lumyongsatien, J.; Bedavanija, A.; Suwanwech, T. Endotyping of chronic rhinosinusitis with and without polyp using transcription factor analysis. Front. Cell. Infect. Microbiol. 2018, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.; Martin, S. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Griffiths, G.M. Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biology. 2007, 23, 495–517. [Google Scholar] [CrossRef] [PubMed]
- Trambas, C.M.; Griffiths, G.M. Delivering the kiss of death. Nat. Immunol. 2003, 4, 399–403. [Google Scholar] [CrossRef]
- Mittrücker, H.-W.; Visekruna, A.; Huber, M. Heterogeneity in the differentiation and function of CD8+ T cells. Arch. Immunol. Et Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Bernstein, J.M.; Ballow, M.; Rich, G.; Allen, C.; Swanson, M.; Dmochowski, J. Lymphocyte subpopulations and cytokines in nasal polyps: Is there a local immune system in the nasal polyp? Otolaryngol.—Head Neck Surg. 2004, 130, 526–535. [Google Scholar] [CrossRef]
- Derycke, L.; Eyerich, S.; Van Crombruggen, K.; Perez-Novo, C.; Holtappels, G.; Deruyck, N.; Gevaert, P.; Bachert, C. Mixed T helper cell signatures in chronic rhinosinusitis with and without polyps. PLoS ONE 2014, 9, e97581. [Google Scholar] [CrossRef]
- Pant, H.; Hughes, A.; Miljkovic, D.; Schembri, M.; Wormald, P.; Macardle, P.; Grose, R.; Zola, H.; Krumbiegel, D. Accumulation of effector memory CD8+ T cells in nasal polyps. Am. J. Rhinol. Allergy 2013, 27, e117–e126. [Google Scholar] [CrossRef]
- Cao, P.-P.; Li, H.-B.; Wang, B.-F.; Wang, S.-B.; You, X.-J.; Cui, Y.-H.; Wang, D.-Y.; Desrosiers, M.; Liu, Z. Distinct immunopathologic characteristics of various types of chronic rhinosinusitis in adult Chinese. J. Allergy Clin. Immunol. 2009, 124, 478–484.e2. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Hulse, K.E.; Tan, B.K.; Schleimer, R.P. B-lymphocyte lineage cells and the respiratory system. J. Allergy Clin. Immunol. 2013, 131, 933–957. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, D. B-cell memory: Are subsets necessary? Nat. Rev. Immunol. 2006, 6, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, R.; Sims, G.P.; Fairhurst, A.-M.; Robbins, R.; da Silva, Y.S.; Spolski, R.; Leonard, W.J.; Lipsky, P.E. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J. Immunol. 2005, 175, 7867–7879. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, K. Clonal selection and learning in the antibody system. Nature 1996, 381, 751–758. [Google Scholar] [CrossRef]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dörner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944. [Google Scholar] [CrossRef]
- Nothelfer, K.; Sansonetti, P.J.; Phalipon, A. Pathogen manipulation of B cells: The best defence is a good offence. Nat. Rev. Microbiol. 2015, 13, 173–184. [Google Scholar] [CrossRef]
- Tan, B.K.; Li, Q.-Z.; Suh, L.; Kato, A.; Conley, D.B.; Chandra, R.K.; Zhou, J.; Norton, J.; Carter, R.; Hinchcliff, M. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2011, 128, 1198–1206.e1. [Google Scholar] [CrossRef]
- Jeffe, J.S.; Seshadri, S.; Hamill, K.J.; Huang, J.H.; Carter, R.; Suh, L.; Hulse, K.E.; Norton, J.; Conley, D.B.; Chandra, R.K. A role for anti-BP180 autoantibodies in chronic rhinosinusitis. Laryngoscope 2013, 123, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-B.; James, L.K.; Davies, A.M.; Wu, Y.-C.B.; Rimmer, J.; Lund, V.J.; Chen, J.-H.; McDonnell, J.M.; Chan, Y.-C.; Hutchins, G.H. Antibodies and superantibodies in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2017, 139, 1195–1204.e11. [Google Scholar] [CrossRef] [PubMed]
- De Schryver, E.; Devuyst, L.; Derycke, L.; Dullaers, M.; Van Zele, T.; Bachert, C.; Gevaert, P. Local immunoglobulin e in the nasal mucosa: Clinical implications. Allergy Asthma Immunol. Res. 2015, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Holtappels, G.; Gevaert, P.; Patou, J.; Dhaliwal, B.; Gould, H.; Bachert, C. Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy 2011, 66, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.D.; Gardam, S.; Gatto, D.; Turner, V.M.; Silke, J.; Brink, R. In vivo control of B-cell survival and antigen-specific B-cell responses. Immunol. Rev. 2010, 237, 90–103. [Google Scholar] [CrossRef]
- Kato, A.; Peters, A.; Suh, L.; Carter, R.; Harris, K.E.; Chandra, R.; Conley, D.; Grammer, L.C.; Kern, R.; Schleimer, R.P. Evidence of a role for B cell–activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2008, 121, 1385–1392.e1382. [Google Scholar] [CrossRef]
- Patadia, M.; Dixon, J.; Conley, D.; Chandra, R.; Peters, A.; Suh, L.A.; Kato, A.; Carter, R.; Harris, K.; Grammer, L. Evaluation of the presence of B-cell attractant chemokines in chronic rhinosinusitis. Am. J. Rhinol. Allergy 2010, 24, 11–16. [Google Scholar] [CrossRef]
- Ahmed, R.; Gray, D. Immunological memory and protective immunity: Understanding their relation. Science 1996, 272, 54–60. [Google Scholar] [CrossRef]
- Miljkovic, D.; Psaltis, A.; Wormald, P.-J.; Vreugde, S. Naive and effector B-cell subtypes are increased in chronic rhinosinusitis with polyps. Am. J. Rhinol. Allergy 2018, 32, 3–6. [Google Scholar] [CrossRef]
- Gevaert, P.; Nouri-Aria, K.T.; Wu, H.; Harper, C.E.; Takhar, P.; Fear, D.J.; Acke, F.; De Ruyck, N.; Banfield, G.; Kariyawasam, H.H. Local receptor revision and class switching to IgE in chronic rhinosinusitis with nasal polyps. Allergy 2013, 68, 55–63. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A. Experimental autoimmune encephalomyelitis induction in genetically B cell–deficient mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.A.; Rosser, E.C.; Mauri, C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res. Ther. 2012, 14, R32. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Zhang, N.; Hellings, P.W.; Bousquet, J. Endotype-driven care pathways in patients with chronic rhinosinusitis. J. Allergy Clin. Immunol. 2018, 141, 1543–1551. [Google Scholar] [CrossRef]
- Ahern, S.; Cervin, A. Inflammation and endotyping in chronic rhinosinusitis—A paradigm shift. Medicina 2019, 55, 95. [Google Scholar] [CrossRef]
- De Greve, G.; Hellings, P.W.; Fokkens, W.J.; Pugin, B.; Steelant, B.; Seys, S.F. Endotype-driven treatment in chronic upper airway diseases. Clin. Transl. Allergy 2017, 7, 22. [Google Scholar] [CrossRef]
- Asano, K.; Ueki, S.; Tamari, M.; Imoto, Y.; Fujieda, S.; Taniguchi, M. Adult-onset eosinophilic airway diseases. Allergy 2020, 75, 3087–3099. [Google Scholar] [CrossRef]
- Cao, P.-P.; Wang, Z.-C.; Schleimer, R.P.; Liu, Z. Pathophysiologic mechanisms of chronic rhinosinusitis and their roles in emerging disease endotypes. Ann. Allergy Asthma Immunol. 2019, 122, 33–40. [Google Scholar] [CrossRef]
- König, K.; Klemens, C.; Haack, M.; Nicoló, M.S.; Becker, S.; Kramer, M.F.; Gröger, M. Cytokine patterns in nasal secretion of non-atopic patients distinguish between chronic rhinosinusitis with or without nasal polys. Allergy Asthma Clin. Immunol. 2016, 12, 19. [Google Scholar] [CrossRef]
- Takabayashi, T.; Kato, A.; Peters, A.T.; Hulse, K.E.; Suh, L.A.; Carter, R.; Norton, J.; Grammer, L.C.; Cho, S.H.; Tan, B.K. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of tissue plasminogen activator expression. Am. J. Respir. Crit. Care Med. 2013, 187, 49–57. [Google Scholar] [CrossRef]
- Bachert, C.; Holtappels, G.; Merabishvili, M.; Meyer, T.; Murr, A.; Zhang, N.; Van Crombruggen, K.; Gevaert, E.; Völker, U.; Bröker, B. Staphylococcus aureus controls interleukin-5 release in upper airway inflammation. J. Proteom. 2018, 180, 53–60. [Google Scholar] [CrossRef]
- Stentzel, S.; Teufelberger, A.; Nordengrün, M.; Kolata, J.; Schmidt, F.; Van Crombruggen, K.; Michalik, S.; Kumpfmüller, J.; Tischer, S.; Schweder, T. Staphylococcal serine protease–like proteins are pacemakers of allergic airway reactions to Staphylococcus aureus. J. Allergy Clin. Immunol. 2017, 139, 492–500.e8. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Pulsipher, A.; Gabrielsen, D.A.; Alt, J.A. Biologics in chronic rhinosinusitis: An update and thoughts for future directions. Am. J. Rhinol. Allergy 2018, 32, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Soler, Z.M.; Sauer, D.; Mace, J.; Smith, T.L. Impact of mucosal eosinophilia and nasal polyposis on quality-of-life outcomes after sinus surgery. Otolaryngol. -Head Neck Surg. 2010, 142, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Matsuwaki, Y.; Ookushi, T.; Asaka, D.; Mori, E.; Nakajima, T.; Yoshida, T.; Kojima, J.; Chiba, S.; Ootori, N.; Moriyama, H. Chronic rhinosinusitis: Risk factors for the recurrence of chronic rhinosinusitis based on 5-year follow-up after endoscopic sinus surgery. Int. Arch. Allergy Immunol. 2008, 146, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Soler, Z.M.; Sauer, D.A.; Mace, J.; Smith, T.L. Relationship between clinical measures and histopathologic findings in chronic rhinosinusitis. Otolaryngol.—Head Neck Surg. 2009, 141, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Van Zele, T.; Holtappels, G.; Gevaert, P.; Bachert, C. Differences in initial immunoprofiles between recurrent and nonrecurrent chronic rhinosinusitis with nasal polyps. Am. J. Rhinol. Allergy 2014, 28, 192–198. [Google Scholar] [CrossRef]
- Gonzalo, J.-A.; Lloyd, C.M.; Wen, D.; Albar, J.P.; Wells, T.N.; Proudfoot, A.; Martinez-a, C.; Dorf, M.; Bjerke, T.; Coyle, A.J. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J. Exp. Med. 1998, 188, 157–167. [Google Scholar] [CrossRef]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The interleukin-12/interleukin-12-receptor system: Role in normal and pathologic immune responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Ellis, T.N.; Beaman, B.L. Interferon-γ activation of polymorphonuclear neutrophil function. Immunology 2004, 112, 2–12. [Google Scholar] [CrossRef]
- Skroza, N.; Proietti, I.; Pampena, R.; La Viola, G.; Bernardini, N.; Nicolucci, F.; Tolino, E.; Zuber, S.; Soccodato, V.; Potenza, C. Correlations between psoriasis and inflammatory bowel diseases. BioMed Res. Int. 2013, 2013, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Eidenschenk, C.; Rutz, S.; Liesenfeld, O.; Ouyang, W. Role of IL-22 in microbial host defense. Interleukin-10 Health Dis. 2014, 380, 213–236. [Google Scholar]
- Venkatesan, N.; Lavigne, P.; Lavigne, F.; Hamid, Q. Effects of fluticasone furoate on clinical and immunological outcomes (IL-17) for patients with nasal polyposis naive to steroid treatment. Ann. Otol. Rhinol. Laryngol. 2016, 125, 213–218. [Google Scholar] [CrossRef]
- Wen, W.; Liu, W.; Zhang, L.; Bai, J.; Fan, Y.; Xia, W.; Luo, Q.; Zheng, J.; Wang, H.; Li, Z. Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J. Allergy Clin. Immunol. 2012, 129, 1522–1528.e1525. [Google Scholar] [CrossRef] [PubMed]
- Calus, L.; Van Bruaene, N.; Bosteels, C.; Dejonckheere, S.; Van Zele, T.; Holtappels, G.; Bachert, C.; Gevaert, P. Twelve-year follow-up study after endoscopic sinus surgery in patients with chronic rhinosinusitis with nasal polyposis. Clin. Transl. Allergy 2019, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, D.W.; Gevaert, P. Chronic rhinosinusitis without nasal polyps. J. Allergy Clin. Immunol. Pract. 2016, 4, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, N.; Bo, M.; Holtappels, G.; Zheng, M.; Lou, H.; Wang, H.; Zhang, L.; Bachert, C. Diversity of TH cytokine profiles in patients with chronic rhinosinusitis: A multicenter study in Europe, Asia, and Oceania. J. Allergy Clin. Immunol. 2016, 138, 1344–1353. [Google Scholar] [CrossRef]
- Delemarre, T.; Holtappels, G.; De Ruyck, N.; Zhang, N.; Nauwynck, H.; Bachert, C.; Gevaert, E. Type 2 inflammation in chronic rhinosinusitis without nasal polyps: Another relevant endotype. J. Allergy Clin. Immunol. 2020, 146, 337–343.e6. [Google Scholar] [CrossRef]
- Smith, T.L.; Litvack, J.R.; Hwang, P.H.; Loehrl, T.A.; Mace, J.C.; Fong, K.J.; James, K.E. Determinants of outcomes of sinus surgery: A multi-institutional prospective cohort study. Otolaryngol.-Head Neck Surg. 2010, 142, 55–63. [Google Scholar] [CrossRef]
- DeConde, A.S.; Mace, J.C.; Levy, J.M.; Rudmik, L.; Alt, J.A.; Smith, T.L. Prevalence of polyp recurrence after endoscopic sinus surgery for chronic rhinosinusitis with nasal polyposis. Laryngoscope 2017, 127, 550–555. [Google Scholar] [CrossRef]
- Succar, E.F.; Li, P.; Ely, K.A.; Chowdhury, N.I.; Chandra, R.K.; Turner, J.H. Neutrophils are underrecognized contributors to inflammatory burden and quality of life in chronic rhinosinusitis. Allergy 2020, 75, 713–716. [Google Scholar] [CrossRef]
- Lou, H.; Meng, Y.; Piao, Y.; Zhang, N.; Bachert, C.; Wang, C.; Zhang, L. Cellular phenotyping of chronic rhinosinusitis with nasal polyps. Rhinol. J. 2018, 54, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Bochner, B.S.; Stevens, W.W. Biology and function of eosinophils in chronic rhinosinusitis with or without nasal polyps. Allergy Asthma Immunol. Res. 2021, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, W.J.; Lund, V.; Luong, A.U.; Orlandi, R.R. A comparison of international guidelines for rhinosinusitis. J. Allergy Clin. Immunol. Pract. 2022, 10, 1418–1422. [Google Scholar] [CrossRef]
- Chong, L.Y.; Head, K.; Hopkins, C.; Philpott, C.; Schilder, A.G.; Burton, M.J. Intranasal steroids versus placebo or no intervention for chronic rhinosinusitis. Cochrane Database Syst. Rev. 2016, 26, CD011996. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Pawankar, R.; Zhang, L.; Bunnag, C.; Fokkens, W.J.; Hamilos, D.L.; Jirapongsananuruk, O.; Kern, R.; Meltzer, E.O.; Mullol, J. ICON: Chronic rhinosinusitis. World Allergy Organ. J. 2014, 7, 25. [Google Scholar] [CrossRef]
- Rudmik, L.; Soler, Z.M. Medical therapies for adult chronic sinusitis: A systematic review. J. Am. Med. Assoc. 2015, 314, 926–939. [Google Scholar] [CrossRef]
- Sharma, R.; Lakhani, R.; Rimmer, J.; Hopkins, C. Surgical interventions for chronic rhinosinusitis with nasal polyps. Cochrane Database Syst. Rev. 2014, 12, CD006991. [Google Scholar] [CrossRef]
- Cardona, V.; Luengo, O.; Labrador-Horrillo, M. Immunotherapy in allergic rhinitis and lower airway outcomes. Allergy 2017, 72, 35–42. [Google Scholar] [CrossRef]
- Mann, C.; Schmidtmann, I.; Bopp, T.; Brieger, J.; Fruth, K. Treg activation and their role in different subtypes of chronic rhinosinusitis. Allergy 2020, 75, 2687–2689. [Google Scholar] [CrossRef]
- Hong, H.; Liao, S.; Chen, F.; Yang, Q.; Wang, D.Y. Role of IL-25, IL-33, and TSLP in triggering united airway diseases toward type 2 inflammation. Allergy 2020, 75, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.G.; Peters, A.T.; Kato, A.; Stevens, W.W. Use of endotypes, phenotypes, and inflammatory markers to guide treatment decisions in chronic rhinosinusitis. Ann. Allergy Asthma Immunol. 2020, 124, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Han, J.K.; Desrosiers, M.; Hellings, P.W.; Amin, N.; Lee, S.E.; Mullol, J.; Greos, L.S.; Bosso, J.V.; Laidlaw, T.M. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): Results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet 2019, 394, 1638–1650. [Google Scholar] [CrossRef]
- Bachert, C.; Sousa, A.R.; Lund, V.J.; Scadding, G.K.; Gevaert, P.; Nasser, S.; Durham, S.R.; Cornet, M.E.; Kariyawasam, H.H.; Gilbert, J. Reduced need for surgery in severe nasal polyposis with mepolizumab: Randomized trial. J. Allergy Clin. Immunol. 2017, 140, 1024–1031.e14. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, T.M.; Prussin, C.; Panettieri, R.A.; Lee, S.; Ferguson, B.J.; Adappa, N.D.; Lane, A.P.; Palumbo, M.L.; Sullivan, M.; Archibald, D. Dexpramipexole depletes blood and tissue eosinophils in nasal polyps with no change in polyp size. Laryngoscope 2019, 129, E61–E66. [Google Scholar] [CrossRef]
- Gevaert, P.; Calus, L.; Van Zele, T.; Blomme, K.; De Ruyck, N.; Bauters, W.; Hellings, P.; Brusselle, G.; De Bacquer, D.; Van Cauwenberge, P. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J. Allergy Clin. Immunol. 2013, 131, 110–116.e1. [Google Scholar] [CrossRef]
- Fokkens, W.J.; Lund, V.; Bachert, C.; Mullol, J.; Bjermer, L.; Bousquet, J.; Canonica, G.W.; Deneyer, L.; Desrosiers, M.; Diamant, Z. EUFOREA consensus on biologics for CRSwNP with or without asthma. Allergy 2019, 74, 2312–2319. [Google Scholar] [CrossRef]
- Kim, C.; Han, J.; Wu, T.; Bachert, C.; Fokkens, W.; Hellings, P.; Hopkins, C.; Lee, S.; Mullol, J.; Lee, J.T. Role of biologics in chronic rhinosinusitis with nasal polyposis: State of the art review. Otolaryngol.–Head Neck Surg. 2021, 164, 57–66. [Google Scholar] [CrossRef]
- Brown, W.C.; Senior, B. A critical look at the efficacy and costs of biologic therapy for chronic rhinosinusitis with nasal polyposis. Curr. Allergy Asthma Rep. 2020, 20, 16. [Google Scholar] [CrossRef]
- Karauzum, H.; Venkatasubramaniam, A.; Adhikari, R.P.; Kort, T.; Holtsberg, F.W.; Mukherjee, I.; Mednikov, M.; Ortines, R.; Nguyen, N.T.; Doan, T. IBT-V02: A multicomponent toxoid vaccine protects against primary and secondary skin infections caused by Staphylococcus aureus. Front. Immunol. 2021, 12, 475. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaghayegh, G.; Cooksley, C.; Ramezanpour, M.; Wormald, P.-J.; Psaltis, A.J.; Vreugde, S. Chronic Rhinosinusitis, S. aureus Biofilm and Secreted Products, Inflammatory Responses, and Disease Severity. Biomedicines 2022, 10, 1362. https://doi.org/10.3390/biomedicines10061362
Shaghayegh G, Cooksley C, Ramezanpour M, Wormald P-J, Psaltis AJ, Vreugde S. Chronic Rhinosinusitis, S. aureus Biofilm and Secreted Products, Inflammatory Responses, and Disease Severity. Biomedicines. 2022; 10(6):1362. https://doi.org/10.3390/biomedicines10061362
Chicago/Turabian StyleShaghayegh, Gohar, Clare Cooksley, Mahnaz Ramezanpour, Peter-John Wormald, Alkis James Psaltis, and Sarah Vreugde. 2022. "Chronic Rhinosinusitis, S. aureus Biofilm and Secreted Products, Inflammatory Responses, and Disease Severity" Biomedicines 10, no. 6: 1362. https://doi.org/10.3390/biomedicines10061362
APA StyleShaghayegh, G., Cooksley, C., Ramezanpour, M., Wormald, P.-J., Psaltis, A. J., & Vreugde, S. (2022). Chronic Rhinosinusitis, S. aureus Biofilm and Secreted Products, Inflammatory Responses, and Disease Severity. Biomedicines, 10(6), 1362. https://doi.org/10.3390/biomedicines10061362