
Discovering Common miRNA Signatures Underlying Female-Specific Cancers via a Machine Learning Approach Driven by the Cancer Hallmark ERBB

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
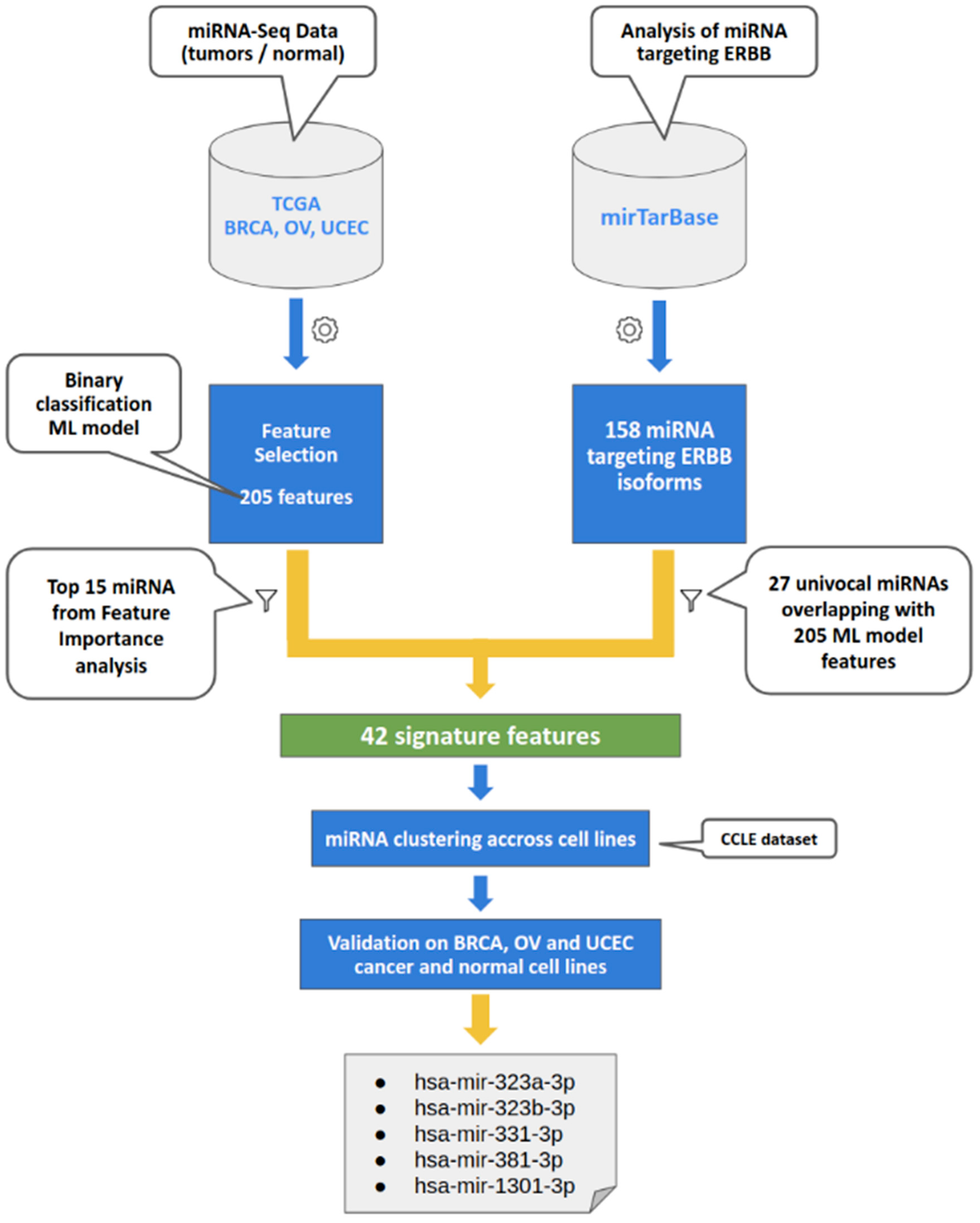
2.1. Data Collection and Machine Learning Analysis
2.2. CCLE Dataset Analyses, Hierarchical Clustering, and Database Explorations
2.3. Cell Culture
2.4. RNA Extraction and Real-Time PCR
3. Results
3.1. Integrated Computational Approaches
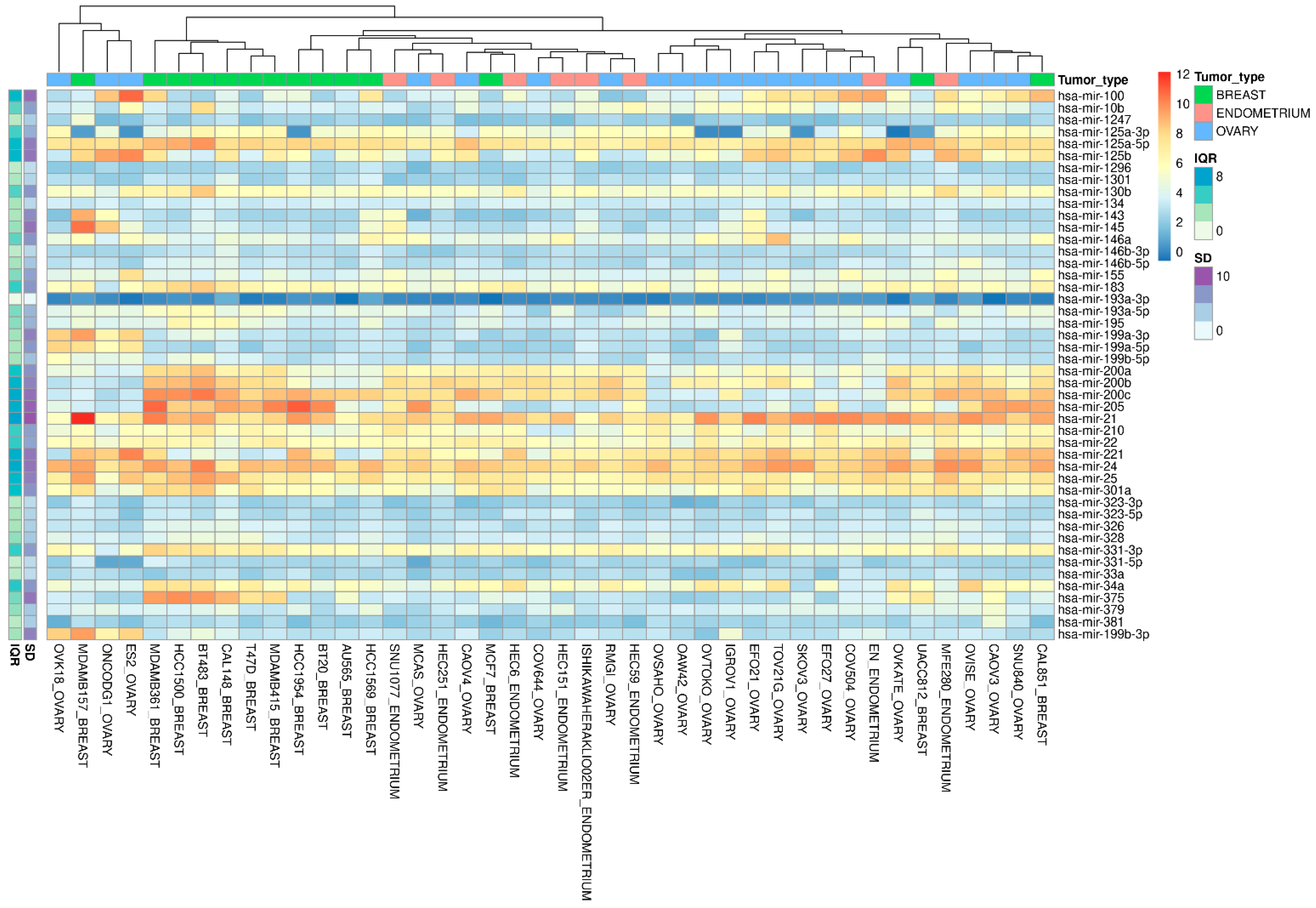
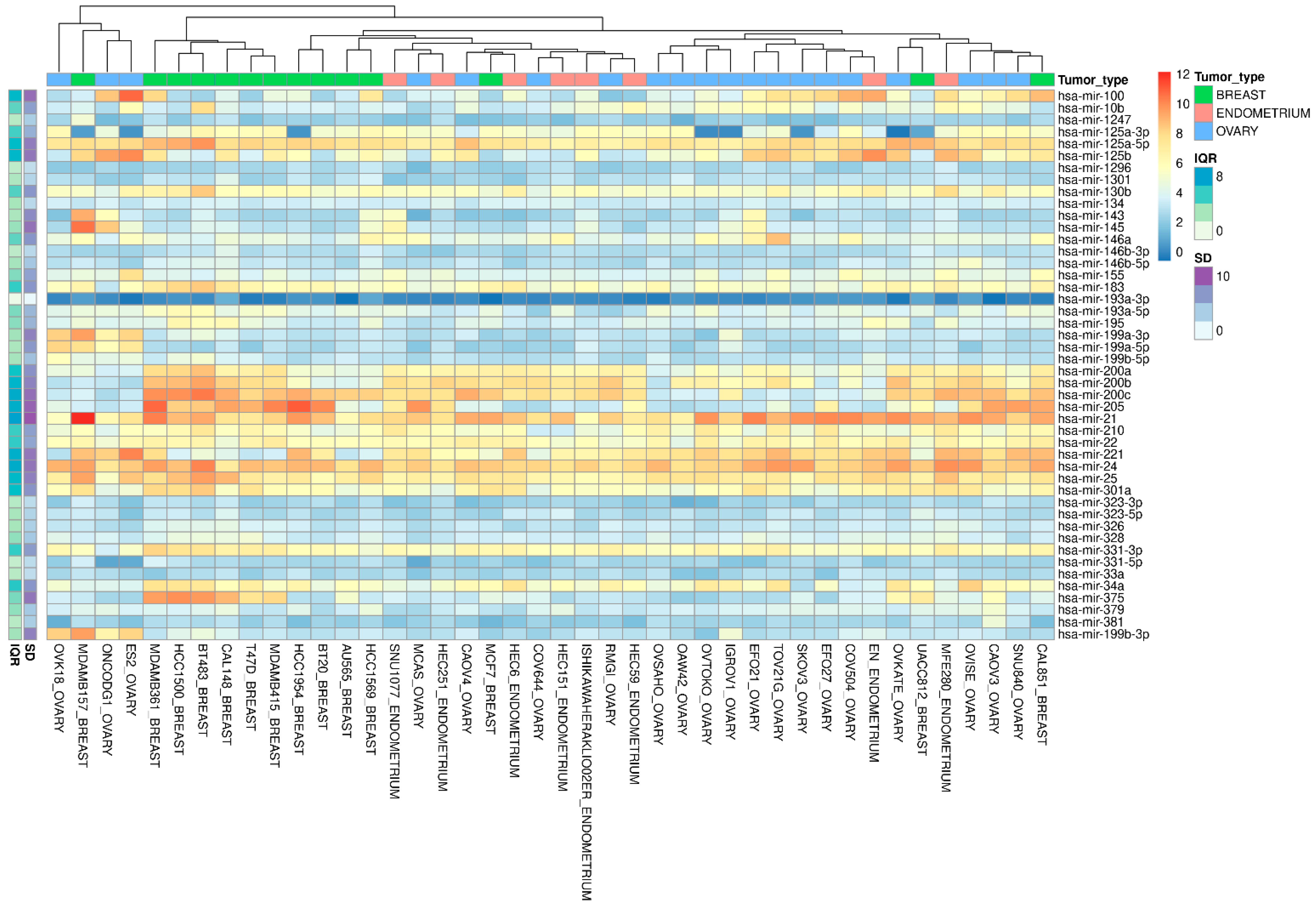
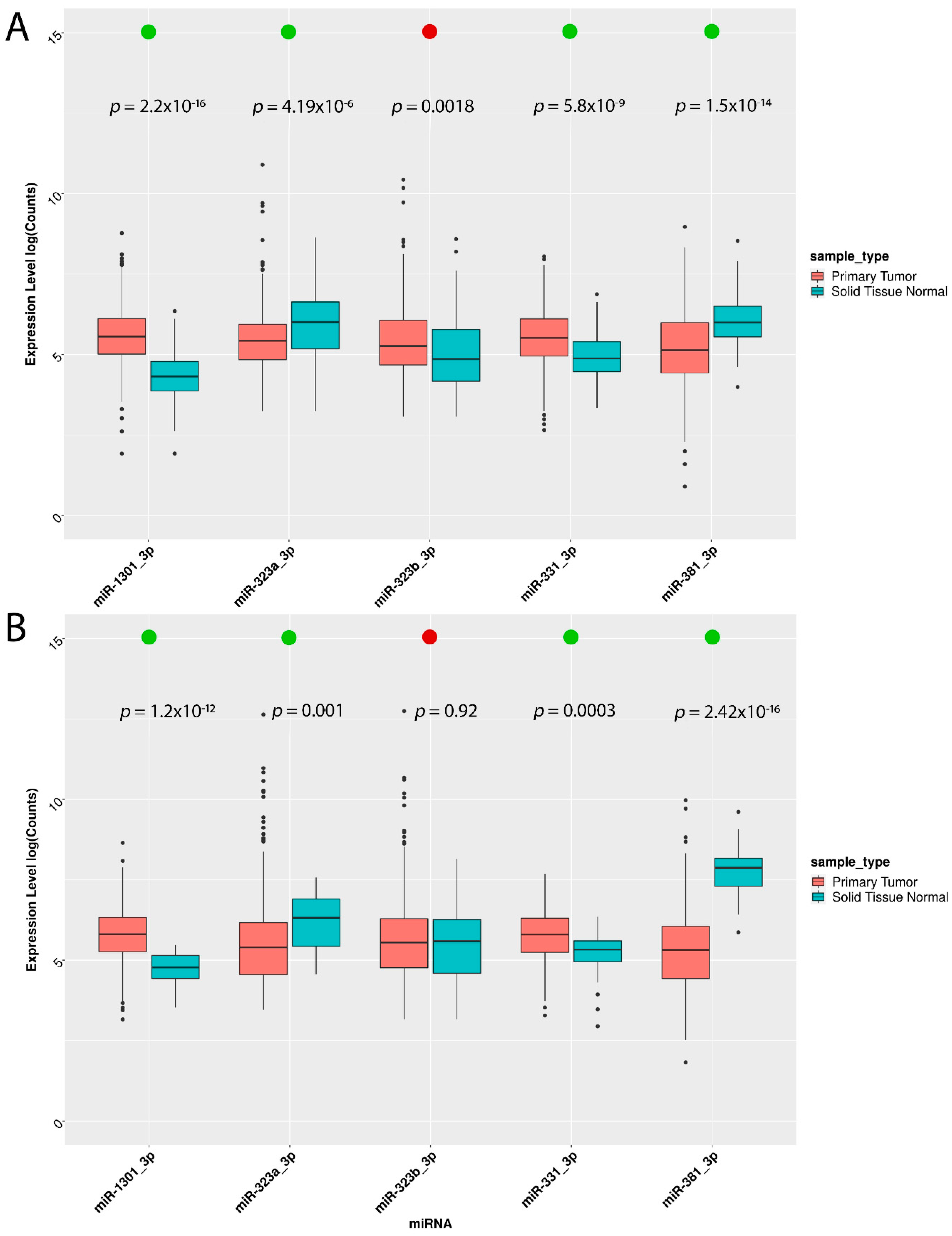
3.2. miRNA Validation in the Cancer Cell Line Encyclopedia
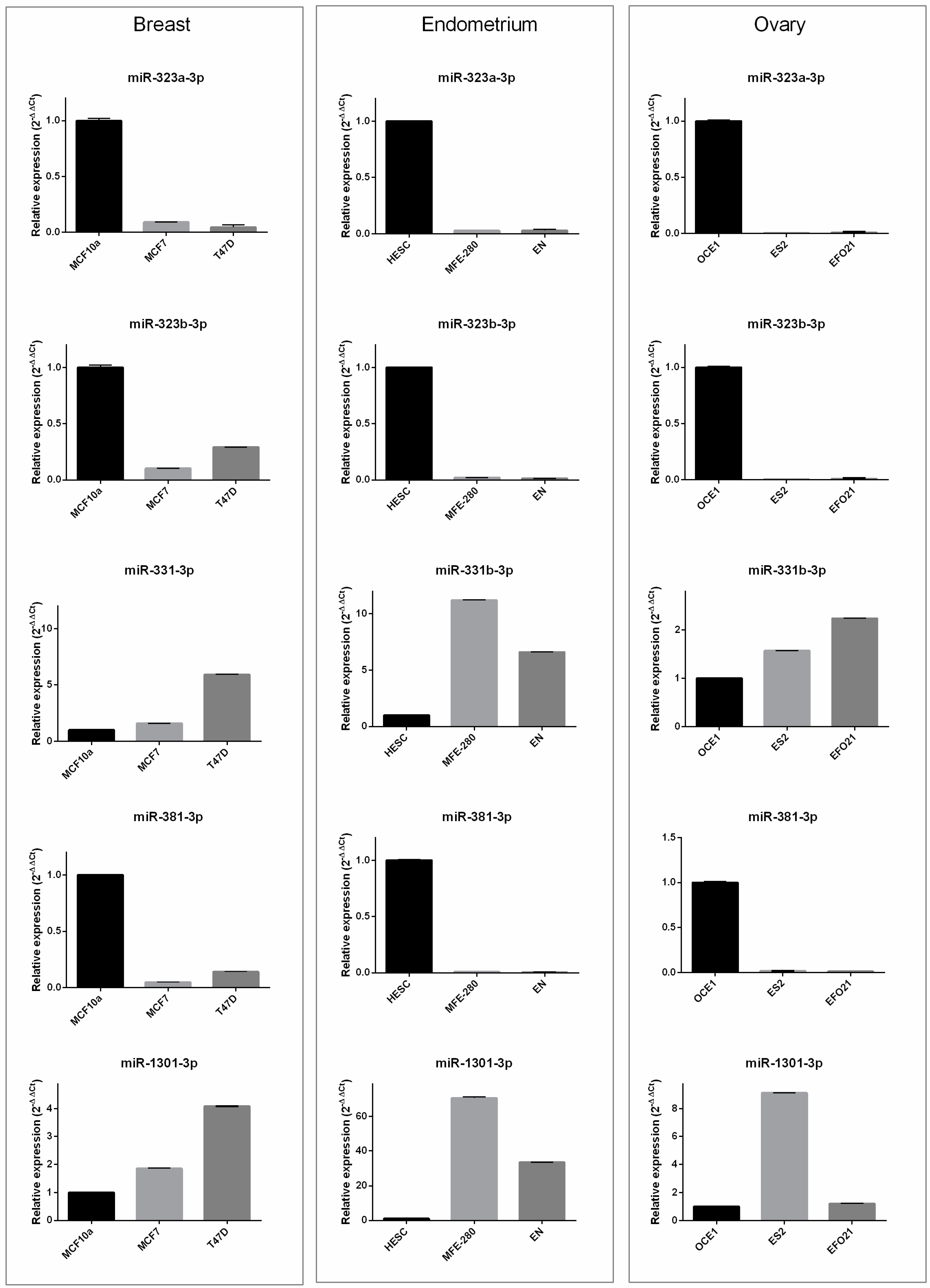
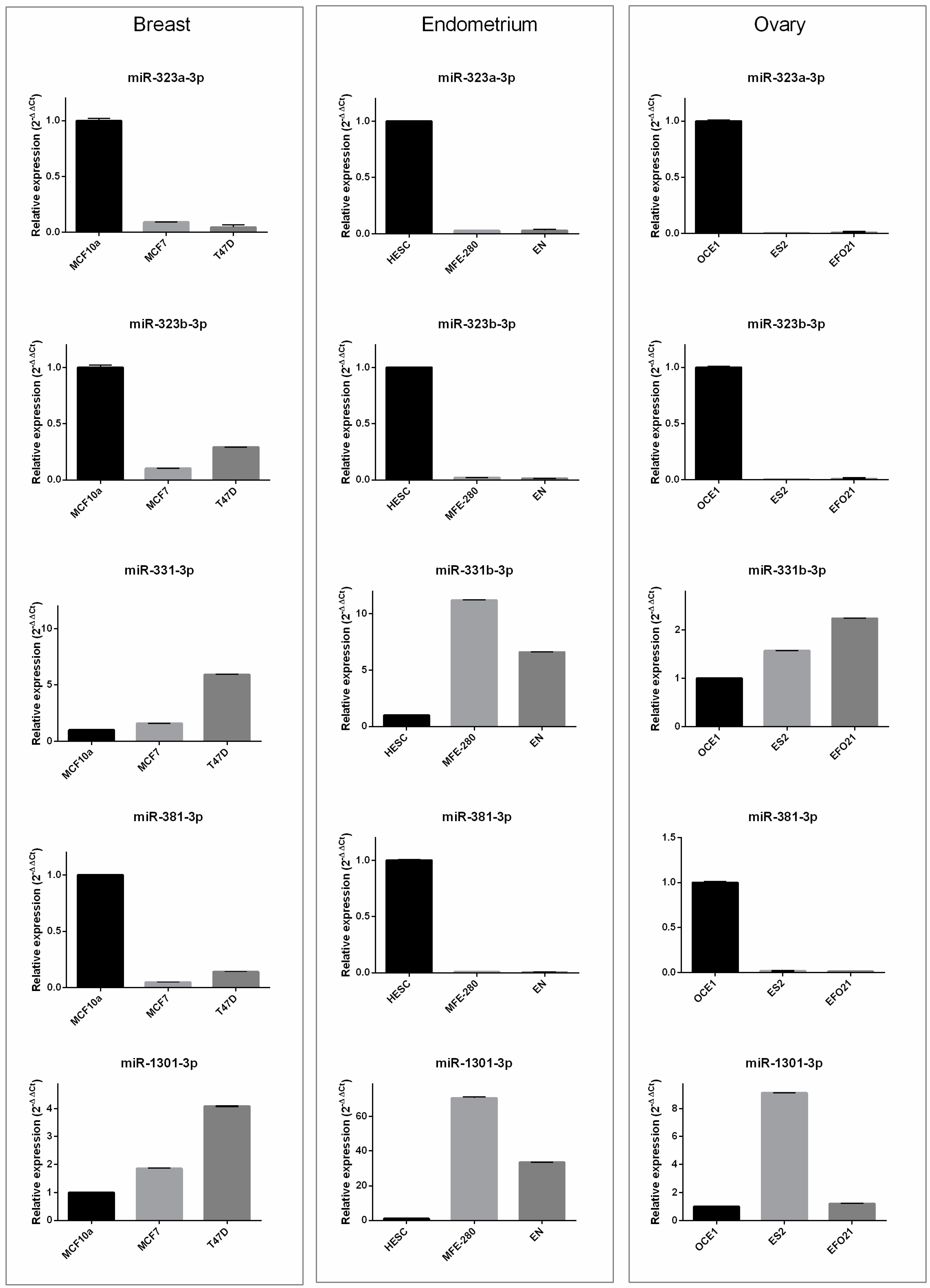
3.3. In Vitro Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fiorica, J.V. Breast Cancer Screening, Mammography, and Other Modalities. Clin. Obstet. Gynecol. 2016, 59, 688–709. [Google Scholar] [CrossRef] [PubMed]
- Barba, D.; León-Sosa, A.; Lugo, P.; Suquillo, D.; Torres, F.; Surre, F.; Trojman, L.; Caicedo, A. Breast cancer, screening and diagnostic tools: All you need to know. Crit. Rev. Oncol. Hematol. 2021, 157, 103174. [Google Scholar] [CrossRef]
- Romanowska, J.; Joshi, A. From genotype to phenotype: Through chromatin. Genes 2019, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, R.; Cavaliere, C.; Soricelli, A.; Salvatore, M.; Pecchia, L.; Franzese, M. Radiomic and genomic machine learning method performance for prostate cancer diagnosis: Systematic literature review. J. Med. Internet Res. 2021, 23, e22394. [Google Scholar] [CrossRef]
- Murali, N.; Kucukkaya, A.; Petukhova, A.; Onofrey, J.; Chapiro, J. Supervised Machine Learning in Oncology: A Clinician’s Guide. Dig. Dis. Interv. 2020, 4, 73–81. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Pane, K.; Affinito, O.; Zanfardino, M.; Castaldo, R.; Incoronato, M.; Salvatore, M.; Franzese, M. An Integrative Computational Approach Based on Expression Similarity Signatures to Identify Protein–Protein Interaction Networks in Female-Specific Cancers. Front. Genet. 2020, 11, 612521. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Zeybek, B.; Santin, A.D.; Fader, A.N. Targeting human epidermal growth factor receptor 2 (HER2) in gynecologic malignancies. Curr. Opin. Obstet. Gynecol. 2020, 32, 57–64. [Google Scholar] [CrossRef]
- Luo, H.; Xu, X.; Ye, M.; Sheng, B.; Zhu, X. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies. PLoS ONE 2018, 13, e0191972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandella, F.M.; Mariniello, R.M.; Iervolino, P.L.C.; Imperlini, E.; Mandola, A.; Verde, A.; De Stefano, A.E.; Pane, K.; Franzese, M.; Esposito, S.; et al. miR-650 promotes motility of anaplastic thyroid cancer cells by targeting PPP2CA. Endocrine 2019, 65, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Pane, K.; Mirabelli, P.; Coppola, L.; Illiano, E.; Salvatore, M.; Franzese, M. New Roadmaps for Non-muscle-invasive Bladder Cancer With Unfavorable Prognosis. Front. Chem. 2020, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Palma, F.; Affinito, A.; Nuzzo, S.; Roscigno, G.; Scognamiglio, I.; Ingenito, F.; Martinez, L.; Franzese, M.; Zanfardino, M.; Soricelli, A.; et al. miR-34c-3p targets CDK1 a synthetic lethality partner of KRAS in non-small cell lung cancer. Cancer Gene Ther. 2021, 28, 413–426. [Google Scholar] [CrossRef]
- Liolios, T.; Kastora, S.L.; Colombo, G. MicroRNAs in Female Malignancies. Cancer Inform. 2019, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, T.; Wang, Y.; Fu, J.; Yang, Q.; Wu, J.; Zhou, H. MiRNA–mRNA associated with survival in endometrial cancer. Front. Genet. 2019, 10, 743. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, L.; Wang, B.; Wei, R.; Wang, Y.; Wan, J.; Zhang, C.; Zhao, L.; Zhu, X.; Zhang, Y.; et al. MiR-337–3p suppresses proliferation of epithelial ovarian cancer by targeting PIK3CA and PIK3CB. Cancer Lett. 2020, 469, 54–67. [Google Scholar] [CrossRef]
- Zhou, W.J.; Zhang, J.; Xie, F.; Wu, J.N.; Ye, J.F.; Wang, J.; Wu, K.; Li, M.Q. CD45RO-CD8+ T cell-derived exosomes restrict estrogen-driven endometrial cancer development via the ERβ/miR-765/PLP2/Notch axis. Theranostics 2021, 11, 5330–5345. [Google Scholar] [CrossRef]
- Riaz, M.; van Jaarsveld, M.T.M.; Hollestelle, A.; Prager-van der Smissen, W.J.C.; Heine, A.A.J.; Boersma, A.W.M.; Liu, J.; Helmijr, J.; Ozturk, B.; Smid, M.; et al. MiRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013, 15, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschfeld, M.; Ge, I.; Rücker, G.; Waldschmidt, J.; Mayer, S.; Jäager, M.; Voigt, M.; Kammerer, B.; Nöthling, C.; Berner, K.; et al. Mutually distinguishing microRNA signatures of breast, ovarian and endometrial cancers in vitro. Mol. Med. Rep. 2020, 22, 4048–4060. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Hirschfeld, M.; Berner, K.; Jaeger, M.; Grundner-Culemann, F.; Schlosser, P.; Asberger, J.; Weiss, D.; Noethling, C.; Mayer, S.; et al. Discovery of potential serum and urine-based microRNA as minimally-invasive biomarkers for breast and gynecological cancer. Cancer Biomark. 2020, 27, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Gupta, S.K.; Vera, J. Computational Biology of Non-Coding RNA Methods and Protocols; Humana Press: Totowa, NJ, USA, 2019; ISBN 9781493989812. [Google Scholar]
- Grimaldi, A.M.; Incoronato, M. Clinical translatability of “identified” circulating mirnas for diagnosing breast cancer: Overview and update. Cancers 2019, 11, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshamrani, A.A. Roles of microRNAs in Ovarian Cancer Tumorigenesis: Two Decades Later, What Have We Learned? Front. Oncol. 2020, 10, 1084. [Google Scholar] [CrossRef]
- Klicka, K.; Grzywa, T.M.; Klinke, A.; Mielniczuk, A.; Włodarski, P.K. The Role of miRNAs in the Regulation of Endometrial Cancer Invasiveness and Metastasis—A Systematic Review. Cancers 2021, 13, 3393. [Google Scholar] [CrossRef]
- Rehman, O.; Zhuang, H.; Ali, A.M.; Ibrahim, A.; Li, Z. Validation of miRNAs as Breast Cancer Biomarkers with a Machine Learning Approach. Cancers 2019, 11, 431. [Google Scholar] [CrossRef]
- Hamidi, F.; Gilani, N.; Belaghi, R.A.; Sarbakhsh, P.; Edgünlü, T.; Santaguida, P. Exploration of Potential miRNA Biomarkers and Prediction for Ovarian Cancer Using Artificial Intelligence. Front. Genet. 2021, 12, 2079. [Google Scholar] [CrossRef]
- Fridrichova, I.; Kalinkova, L.; Karhanek, M.; Smolkova, B.; Machalekova, K.; Wachsmannova, L.; Nikolaieva, N.; Kajo, K. miR-497-5p Decreased Expression Associated with High-Risk Endometrial Cancer. Int. J. Mol. Sci. 2020, 22, 127. [Google Scholar] [CrossRef]
- Zheng, K.; You, Z.H.; Wang, L.; Zhou, Y.; Li, L.P.; Li, Z.W. MLMDA: A machine learning approach to predict and validate MicroRNA-disease associations by integrating of heterogenous information sources. J. Transl. Med. 2019, 17, 260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zheng, J.; Li, L.; Huang, R.; Ren, H.; Wang, D.; Dai, Z.; Su, X. Application of Machine Learning Algorithms to Predict Central Lymph Node Metastasis in T1-T2, Non-invasive, and Clinically Node Negative Papillary Thyroid Carcinoma. Front. Med. 2021, 8, 635771. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Zhuang, H.; Ibrahim, A.; Rehman, O.; Huang, M.; Wu, A. A machine learning approach for the classification of kidney cancer subtypes using miRNA genome data. Appl. Sci. 2018, 8, 2422. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, A.; Matsuzaki, J.; Yamamoto, Y.; Yoneoka, Y.; Takahashi, K.; Shimizu, H.; Uehara, T.; Ishikawa, M.; Ikeda, S.I.; Sonoda, T.; et al. Integrated extracellular microRNA profiling for ovarian cancer screening. Nat. Commun. 2018, 9, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Z.; Domeniconi, C.; Zhang, X.; Yu, G. Cooperative driver pathway discovery via fusion of multi-relational data of genes, miRNAs and pathways. Brief. Bioinform. 2021, 22, 1984–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. Off. Publ. Indian Soc. Dent. Res. 2017, 28, 687–694. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. MiRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Jiang, H.; Gu, J.; Du, J.; Qi, X.; Qian, C.; Fei, B. A 21-gene Support Vector Machine classifier and a 10-gene risk score system constructed for patients with gastric cancer. Mol. Med. Rep. 2020, 21, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Hase, T.; Li, H.P.; Prabhakar, S.; Kitano, H.; Ng, S.K.; Ghosh, S.; Wee, L.J.K. A machine learning approach for the identification of key markers involved in brain development from single-cell transcriptomic data. BMC Genom. 2016, 17, 1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Lai, Y.; Xu, D.; Xu, L.; Song, L.; Zhou, J.; Song, C.; Wang, J. Diagnosis of thyroid neoplasm using support vector machine algorithms based on platelet RNA-seq. Endocrine 2021, 72, 758–783. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523. [Google Scholar] [CrossRef]
- Lyu, H.; Huang, J.; Edgerton, S.M.; Thor, A.D.; He, Z.; Liu, B. Increased erbB3 promotes erbB2/neu-driven mammary tumor proliferation and co-targeting of erbB2/erbB3 receptors exhibits potent inhibitory effects on breast cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6143–6156. [Google Scholar]
- Meher, P.K.; Begam, S.; Sahu, T.K.; Gupta, A.; Kumar, A.; Kumar, U.; Rao, A.R.; Singh, K.P.; Dhankher, O.P. ASRmiRNA: Abiotic Stress-Responsive miRNA Prediction in Plants by Using Machine Learning Algorithms with Pseudo K-Tuple Nucleotide Compositional Features. Int. J. Mol. Sci. 2022, 23, 1612. [Google Scholar] [CrossRef]
- Yerukala Sathipati, S.; Ho, S.-Y. Novel miRNA signature for predicting the stage of hepatocellular carcinoma. Sci. Rep. 2020, 10, 14452. [Google Scholar] [CrossRef]
- Dong, J.; Xu, M. A 19-miRNA Support Vector Machine classifier and a 6-miRNA risk score system designed for ovarian cancer patients. Oncol. Rep. 2019, 41, 3233–3243. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, S.; Yazdanparast, A.; Li, M.; Pawar, A.V.; Liu, Y.; Inavolu, S.M.; Cheng, L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genom. 2016, 17, 2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvadores, M.; Fuster-Tormo, F.; Supek, F. Matching cell lines with cancer type and subtype of origin via mutational, epigenomic, and transcriptomic patterns. Sci. Adv. 2020, 6, eaba1862. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Frederick, L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally. Cancer Cell 2009, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Park, C.; Park, S. PMAMCA: Prediction of microRNA-disease association utilizing a matrix completion approach. BMC Syst. Biol. 2019, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; He, M.; Yin, Y.; Yan, T.; Cheng, W.; Huang, Z.; Zhang, L.; Zhang, H.; Liu, P.; Zhu, W.; et al. miR-1296-5p decreases ERBB2 expression to inhibit the cell proliferation in ERBB2-positive breast cancer. Cancer Cell Int. 2017, 17, 95. [Google Scholar] [CrossRef] [Green Version]
- Phan, B.; Majid, S.; Ursu, S.; De Semir, D.; Nosrati, M.; Bezrookove, V.; Kashani-Sabet, M.; Dar, A.A. Tumor suppressor role of microRNA-1296 in triple-negative breast cancer. Oncotarget 2016, 7, 19519. [Google Scholar] [CrossRef] [Green Version]
- Albakr, L.; Alqahtani, F.Y.; Aleanizy, F.S.; Alomrani, A.; Badran, M.; Alhindas, H.; Al-Mohanna, F. Improved delivery of miR-1296 loaded cationic nanoliposomes for effective suppression of triple negative breast cancer. Saudi Pharm. J. 2021, 29, 446–455. [Google Scholar] [CrossRef]
- Zhou, Y.; Ma, G.; Peng, S.; Tuo, M.; Li, Y.; Qin, X.; Yu, Q.; Kuang, S.; Cheng, H.; Li, J. Circ_0000520 contributes to triple-negative breast cancer progression through mediating the miR-1296/ZFX axis. Thorac. Cancer 2021, 12, 2427–2438. [Google Scholar] [CrossRef]
- Lopez-Rincon, A.; Martinez-Archundia, M.; Martinez-Ruiz, G.U.; Schoenhuth, A.; Tonda, A. Automatic discovery of 100-miRNA signature for cancer classification using ensemble feature selection. BMC Bioinform. 2019, 20, 480. [Google Scholar] [CrossRef] [Green Version]
- Leivonen, S.; Sahlberg, K.; Mäkelä, R.; Due, E.; Kallioniemi, O.; Børresen-Dale, A.; Perälä, M. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol. Oncol. 2014, 8, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, E.I.; Papachristopoulou, G.; Ardavanis, A.; Scorilas, A. A comprehensive clinicopathological evaluation of the differential expression of microRNA-331 in breast tumors and its diagnostic significance. Clin. Biochem. 2018, 60, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Epis, M.R.; Giles, K.M.; Barker, A.; Kendrick, T.S.; Leedman, P.J. miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J. Biol. Chem. 2009, 284, 24696–24704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-H.; Sun, M.; Nie, F.-Q.; Ge, Y.-B.; Zhang, E.-B.; Yin, D.-D.; Kong, R.; Xia, R.; Lu, K.-H.; Li, J.-H.; et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol. Cancer 2014, 13, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Sui, Y.; Zheng, X. MiR-331-3p inhibits proliferation and promotes apoptosis by targeting HER2 through the PI3K/Akt and ERK1/2 pathways in colorectal cancer. Oncol. Rep. 2016, 35, 1075–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-Z.; Mu, Q.; Ren, Q.; Xie, L.-J.; Wang, Q.-T.; Wang, C.-P. miR-381-3p suppresses breast cancer progression by inhibition of epithelial–mesenchymal transition. World J. Surg. Oncol. 2021, 19, 230. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Zhang, J.; Li, X.; Li, W.; Li, H.; Fu, P. Long non-coding RNA NORAD inhibition upregulates microRNA-323a-3p to suppress tumorigenesis and development of breast cancer through the PUM1/eIF2 axis. Cell Cycle 2021, 20, 1295–1307. [Google Scholar] [CrossRef]
- Escuin, D.; López-Vilaró, L.; Mora, J.; Bell, O.; Moral, A.; Pérez, I.; Arqueros, C.; García-Valdecasas, B.; Cajal, T.R.Y.; Lerma, E.; et al. Circulating microRNAs in Early Breast Cancer Patients and Its Association With Lymph Node Metastases. Front. Oncol. 2021, 11, 627811. [Google Scholar] [CrossRef]
- Peng, X.; Yan, B.; Shen, Y. MiR-1301-3p inhibits human breast cancer cell proliferation by regulating cell cycle progression and apoptosis through directly targeting ICT1. Breast Cancer 2018, 25, 742–752. [Google Scholar] [CrossRef]
- Douzas, G.; Bacao, F. Self-Organizing Map Oversampling (SOMO) for imbalanced data set learning. Expert Syst. Appl. 2017, 82, 40–52. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Rank | miRNA ID | Importance Weighting | Feature Source a | FC (BRCA) | FC (UCEC) |
|---|---|---|---|---|---|
| 1 | hsa-mir-183 | 100 | Top 15 | 8.49 | 23.62 |
| 2 | hsa-mir-139 | 93.87 | Top 15 | −7.03 | −11.46 |
| 3 | hsa-mir-145 | 89.75 | Top 15, Targeting ERBB | −5.05 | −10.80 |
| 4 | hsa-mir-10b | 85.99 | Top 15 | −3.20 | −6.57 |
| 5 | hsa-mir-337 | 84.56 | Top 15 | −3.72 | −3.01 |
| 6 | hsa-mir-200c | 83.26 | Top 15 | 3.11 | 3.38 |
| 7 | hsa-mir-200a | 82.17 | Top 15 | 4.72 | 6.26 |
| 8 | hsa-mir-100 | 81.09 | Top 15 | −2.93 | −11.61 |
| 9 | hsa-mir-1247 | 80.4 | Top 15 | −2.60 | −11.19 |
| 10 | hsa-mir-195 | 78.89 | Top 15 | −2.33 | −6.27 |
| 11 | hsa-mir-379 | 77.99 | Top 15 | −1.96 | −4.86 |
| 12 | hsa-mir-1301 | 77.47 | Top 15 | 3.59 | 3.36 |
| 13 | hsa-mir-210 | 76.05 | Top 15 | 7.42 | 6.79 |
| 14 | hsa-mir-200b | 75.21 | Top 15 | 3.36 | 4.50 |
| 15 | hsa-mir-381 | 75.13 | Top 15 | −2.05 | −7.24 |
| 16 | hsa-mir-143 | 71.74 | Targeting ERBB | −1.83 | −11.42 |
| 18 | hsa-mir-130b | 70.99 | Targeting ERBB | 2.65 | 3.78 |
| 27 | hsa-mir-3127 | 59.62 | Targeting ERBB | 2.37 | 1.95 |
| 30 | hsa-mir-125b-1 | 57.64 | Targeting ERBB | −3.22 | −5.32 |
| 40 | hsa-mir-331 | 52.59 | Targeting ERBB | 1.94 | 1.65 |
| 43 | hsa-mir-134 | 50.36 | Targeting ERBB | −1.59 | −2.31 |
| 44 | hsa-mir-155 | 50.25 | Targeting ERBB | 2.72 | 2.28 |
| 54 | hsa-mir-199b | 46.53 | Targeting ERBB | 1.01 | −3.24 |
| 58 | hsa-mir-199a-1 | 45.39 | Targeting ERBB | 1.10 | −3.35 |
| 76 | hsa-mir-22 | 41.32 | Targeting ERBB | −1.40 | −1.83 |
| 107 | hsa-mir-21 | 32.12 | Targeting ERBB | 5.01 | 1.03 |
| 108 | hsa-mir-375 | 31.5 | Targeting ERBB | 7.51 | 2.39 |
| 113 | hsa-mir-146b | 29.65 | Targeting ERBB | 1.46 | 1.10 |
| 114 | hsa-mir-326 | 29.11 | Targeting ERBB | −2.25 | −1.14 |
| 141 | hsa-mir-301a | 21.2 | Targeting ERBB | 3.32 | 1.90 |
| 147 | hsa-mir-33a | 18.69 | Targeting ERBB | 2.67 | −1.51 |
| 152 | hsa-mir-323b | 16.72 | Targeting ERBB | 1.19 | 3.21 |
| 154 | hsa-mir-193a | 16.39 | Targeting ERBB | −2.02 | −1.58 |
| 156 | hsa-mir-205 | 15.28 | Targeting ERBB | −2.67 | 51.59 |
| 157 | hsa-mir-25 | 15.11 | Targeting ERBB | −1.04 | −1.01 |
| 162 | hsa-mir-328 | 13.21 | Targeting ERBB | −1.87 | −1.95 |
| 168 | hsa-mir-125a | 10.71 | Targeting ERBB | −1.31 | −2.85 |
| 175 | hsa-mir-221 | 8.38 | Targeting ERBB | 1.03 | −3.12 |
| 180 | hsa-mir-146a | 7.77 | Targeting ERBB | 1.51 | 2.70 |
| 185 | hsa-mir-34a | 6.75 | Targeting ERBB | 1.21 | −1.23 |
| 196 | hsa-mir-24-1 | 2.36 | Targeting ERBB | 1.03 | −1.54 |
| 205 | hsa-mir-1296 | 0.11 | Targeting ERBB | −1.44 | −1.27 |
| Mature_Acc | Mature_ID | Mature_Seq |
|---|---|---|
| MIMAT0005899 | hsa-miR-1247-5p | ACCCGUCCCGUUCGUCCCCGGA |
| MIMAT0022721 | hsa-miR-1247-3p | CCCCGGGAACGUCGAGACUGGAGC |
| MIMAT0005797 | hsa-miR-1301-3p | UUGCAGCUGCCUGGGAGUGACUUC |
| MIMAT0000736 | hsa-miR-381-3p | UAUACAAGGGCAAGCUCUCUGU |
| MIMAT0004700 | hsa-miR-331-5p | CUAGGUAUGGUCCCAGGGAUCC |
| MIMAT0000760 | hsa-miR-331-3p | GCCCCUGGGCCUAUCCUAGAA |
| MIMAT0002809 | hsa-miR-146b-5p | UGAGAACUGAAUUCCAUAGGCU |
| MIMAT0004766 | hsa-miR-146b-3p | UGCCCUGUGGACUCAGUUCUGG |
| MIMAT0004506 | hsa-miR-33a-3p | CAAUGUUUCCACAGUGCAUCAC |
| MIMAT0004614 | hsa-miR-193a-5p | UGGGUCUUUGCGGGCGAGAUGA |
| MIMAT0000459 | hsa-miR-193a-3p | AACUGGCCUACAAAGUCCCAGU |
| MIMAT0005794 | hsa-miR-1296-5p | UUAGGGCCCUGGCUCCAUCUCC |
| MIMAT0015050 | hsa-miR-323b-3p a | CCCAAUACACGGUCGACCUCUU |
| MIMAT0000755 | hsa-miR-323a-3p a | CACAUUACACGGUCGACCUCU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pane, K.; Zanfardino, M.; Grimaldi, A.M.; Baldassarre, G.; Salvatore, M.; Incoronato, M.; Franzese, M. Discovering Common miRNA Signatures Underlying Female-Specific Cancers via a Machine Learning Approach Driven by the Cancer Hallmark ERBB. Biomedicines 2022, 10, 1306. https://doi.org/10.3390/biomedicines10061306
Pane K, Zanfardino M, Grimaldi AM, Baldassarre G, Salvatore M, Incoronato M, Franzese M. Discovering Common miRNA Signatures Underlying Female-Specific Cancers via a Machine Learning Approach Driven by the Cancer Hallmark ERBB. Biomedicines. 2022; 10(6):1306. https://doi.org/10.3390/biomedicines10061306
Chicago/Turabian StylePane, Katia, Mario Zanfardino, Anna Maria Grimaldi, Gustavo Baldassarre, Marco Salvatore, Mariarosaria Incoronato, and Monica Franzese. 2022. "Discovering Common miRNA Signatures Underlying Female-Specific Cancers via a Machine Learning Approach Driven by the Cancer Hallmark ERBB" Biomedicines 10, no. 6: 1306. https://doi.org/10.3390/biomedicines10061306
APA StylePane, K., Zanfardino, M., Grimaldi, A. M., Baldassarre, G., Salvatore, M., Incoronato, M., & Franzese, M. (2022). Discovering Common miRNA Signatures Underlying Female-Specific Cancers via a Machine Learning Approach Driven by the Cancer Hallmark ERBB. Biomedicines, 10(6), 1306. https://doi.org/10.3390/biomedicines10061306