
Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics

 , ,
, ,  , ,
, , 
Abstract
1. Introduction
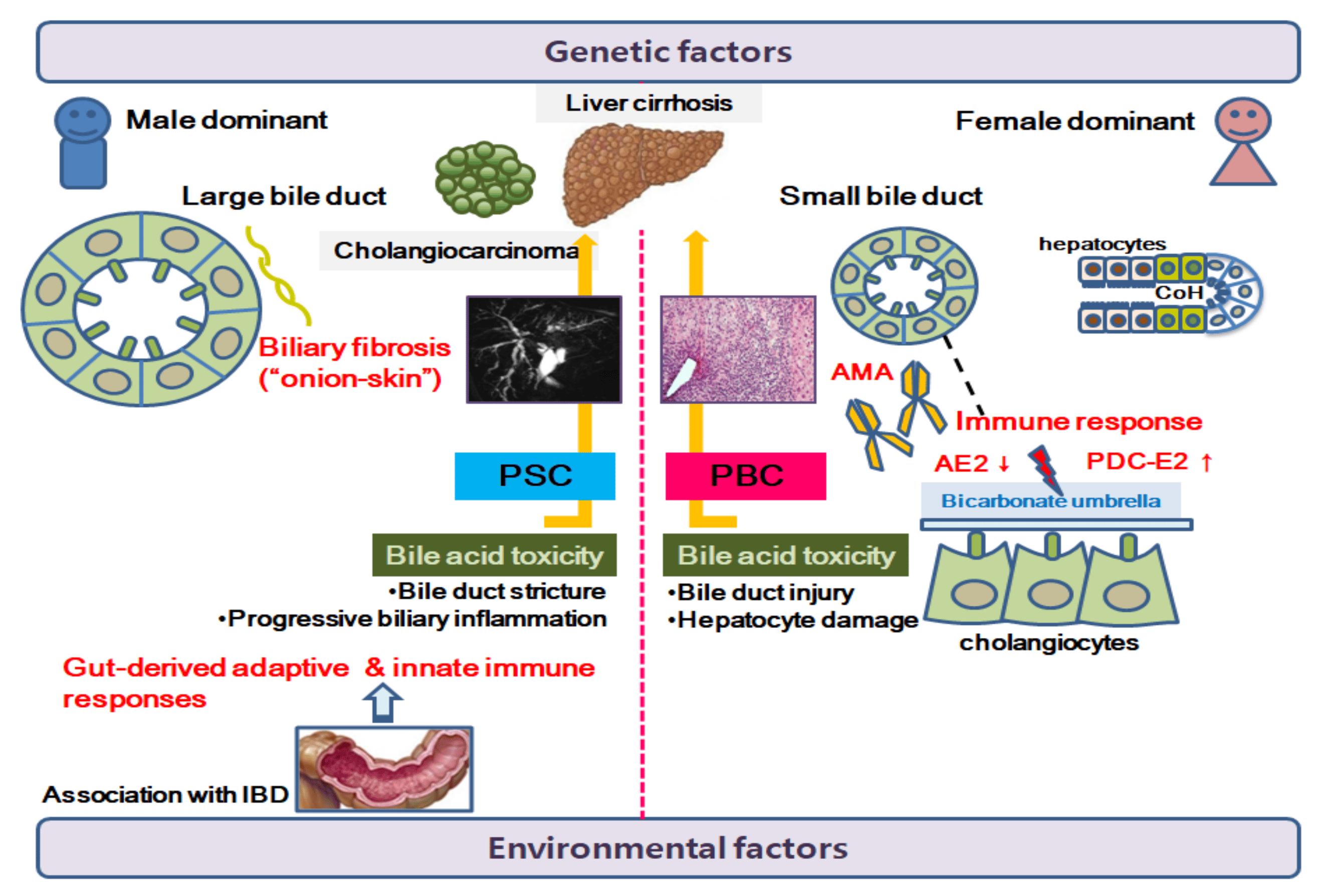
2. Cholangiocyte Pathobiology
3. Primary Biliary Cholangitis
3.1. Pathogenesis of PBC
3.1.1. Genetic Factor
3.1.2. Anion Exchanger Deficiency in PBC
3.1.3. Anti-Mitochondrial Antibodies (AMA) in PBC
3.1.4. Immune Response in PBC
3.1.5. Gut Microbial Profile in PBC
3.2. Currently Approved Disease-Modifying Therapies for PBC
3.2.1. UDCA
3.2.2. Obeticholic Acid (OCA)
3.3. Novel Therapies Currently under Investigation for PBC
3.3.1. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists
3.3.2. Budesonide
3.3.3. Fibroblast Growth Factor (FGF) 19 Analog
3.3.4. Other Farnesoid X Receptor Agonists
3.3.5. Baricitinib
3.3.6. S-adenosyl-L-methionine (SAMe)
3.3.7. Probiotics
3.3.8. Mesenchymal Stem Cells
3.3.9. Anti-Fractalking Antibody E6011
4. Primary Sclerosing Cholangitis
4.1. Mechanism of Chronic Bile Duct Injury in the Bile Duct in PSC
4.1.1. Genetics of PSC
4.1.2. Bile Acid Toxicity to Cholangiocytes and Hepatocytes
4.1.3. Fibrosis Development Related to Cholangiocyte and Hepatic Stellate Cell/Portal Myofibroblast
4.1.4. Gut-Liver Immunity of PSC
4.1.5. Gut Microbial Profile in PSC
4.2. Symptomatic Treatment and Biliary Complications of PSC
4.2.1. Pruritus
4.2.2. Bacterial Cholangitis and Dominant Stricture
4.2.3. Dominant Stricture
4.2.4. Cholangiocarcinoma
4.2.5. Gallbladder Carcinoma
4.2.6. Liver Cirrhosis and Hepatocellular Carcinoma, and Liver Transplantation
4.2.7. Inflammatory Bowel Disease and Colorectal Cancer
4.3. Combination Treatment and Predicting Therapies for PSC
4.3.1. UDCA
4.3.2. FXR Agonist
4.3.3. PPAR Agonist
4.3.4. 3-Hydroxy-3-methylglutaryl Coenzyme A (HMG-CoA) Reductase Inhibitor
4.3.5. Oral Vancomycin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alvaro, D.; Bragazzi, M.C.; Ridola, L. Inflammatory and neoplastic cholangiopathies. Recenti Progress. Med. 2018, 109, 595–599. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Beuers, U.; Ponsioen, C.Y. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: A systematic review. J. Hepatol. 2012, 56, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takikawa, H.; Mochida, S.; Koike, K.; Miwa, H.; Shimosegawa, T. Changing Nomenclature for PBC from “Primary Biliary Cirrhosis” to “Primary Biliary Cholangitis”. J. Gastroenterol. 2016, 51, 748–749. [Google Scholar] [CrossRef]
- Juran, B.D.; Lazaridis, K.N. Environmental factors in primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.J.; Hirschfield, G.M. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J. Autoimmun. 2016, 66, 25–39. [Google Scholar] [CrossRef]
- Bianchi, I.; Carbone, M.; Lleo, A.; Invernizzi, P. Genetics and epigenetics of primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 255–264. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.W.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599. [Google Scholar] [CrossRef]
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013, 58, 2045–2055. [Google Scholar] [CrossRef]
- Sclair, S.N.; Little, E.; Levy, C. Current Concepts in Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis. Clin. Transl. Gastroenterol. 2015, 6, e109. [Google Scholar] [CrossRef]
- Marchioni Beery, R.M.; Vaziri, H.; Forouhar, F. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: A Review Featuring a Women’s Health Perspective. J. Clin. Transl. Hepatol. 2014, 2, 266–284. [Google Scholar] [CrossRef]
- Saxena, R.; Theise, N. Canals of Hering: Recent insights and current knowledge. Semin. Liver Dis. 2004, 24, 43–48. [Google Scholar] [CrossRef]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Banales, J.M.; Prieto, J.; Medina, J.F. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J. Gastroenterol. 2006, 12, 3496–3511. [Google Scholar] [CrossRef]
- Hundt, M.; Basit, H.; John, S. Physiology, Bile Secretion. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Han, Y.; Glaser, S.; Meng, F.; Francis, H.; Marzioni, M.; McDaniel, K.; Alvaro, D.; Venter, J.; Carpino, G.; Onori, P.; et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp. Biol. Med. 2013, 238, 549–565. [Google Scholar] [CrossRef]
- Kanno, N.; LeSage, G.; Glaser, S.; Alvaro, D.; Alpini, G. Functional heterogeneity of the intrahepatic biliary epithelium. Hepatology 2000, 31, 555–561. [Google Scholar] [CrossRef]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and structural features of cholangiocytes in health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef]
- Franchitto, A.; Onori, P.; Renzi, A.; Carpino, G.; Mancinelli, R.; Alvaro, D.; Gaudio, E. Recent advances on the mechanisms regulating cholangiocyte proliferation and the significance of the neuroendocrine regulation of cholangiocyte pathophysiology. Ann. Transl. Med. 2013, 1, 27. [Google Scholar] [CrossRef]
- Sell, S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001, 33, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Juran, B.D.; Boe, G.M.; Slusser, J.P.; de Andrade, M.; Homburger, H.A.; Ghosh, K.; Dickson, E.R.; Lindor, K.D.; Petersen, G.M. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology 2007, 46, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Gulamhusein, A.F.; Lazaridis, K.N. Primary biliary cholangitis, DNA, and beyond: The Relative contribution of genes. Hepatology 2018, 68, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Mungall, A.J.; Palmer, S.A.; Sims, S.K.; Edwards, C.A.; Ashurst, J.L.; Wilming, L.; Jones, M.C.; Horton, R.; Hunt, S.E.; Scott, C.E.; et al. The DNA sequence and analysis of human chromosome 6. Nature 2003, 425, 805–811. [Google Scholar] [CrossRef]
- Joshita, S.; Umemura, T.; Tanaka, E.; Ota, M. Genetics and epigenetics in the pathogenesis of primary biliary cholangitis. Clin. J. Gastroenterol. 2018, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Baragiotta, A.; Heneghan, M.A.; Floreani, A.; Venturi, C.; Underhill, J.A.; Jones, D.E.; James, O.F.; Bassendine, M.F. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: A large-scale study. Hepatology 2006, 44, 667–674. [Google Scholar] [CrossRef]
- Invernizzi, P.; Selmi, C.; Poli, F.; Frison, S.; Floreani, A.; Alvaro, D.; Almasio, P.; Rosina, F.; Marzioni, M.; Fabris, L.; et al. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: A multicenter study of 664 patients and 1992 healthy controls. Hepatology 2008, 48, 1906–1912. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef]
- Cordell, H.J.; Han, Y.; Mells, G.F.; Li, Y.; Hirschfield, G.M.; Greene, C.S.; Xie, G.; Juran, B.D.; Zhu, D.; Qian, D.C.; et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat. Commun. 2015, 6, 8019. [Google Scholar] [CrossRef]
- Poupon, R.; Ping, C.; Chretien, Y.; Corpechot, C.; Chazouilleres, O.; Simon, T.; Heath, S.C.; Matsuda, F.; Poupon, R.E.; Housset, C.; et al. Genetic factors of susceptibility and of severity in primary biliary cirrhosis. J. Hepatol. 2008, 49, 1038–1045. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Ma, X. Epigenetics of Primary Biliary Cholangitis. Adv. Exp. Med. Biol. 2020, 1253, 259–283. [Google Scholar] [CrossRef]
- Lleo, A.; Zhang, W.; Zhao, M.; Tan, Y.; Bernuzzi, F.; Zhu, B.; Liu, Q.; Tan, Q.; Malinverno, F.; Valenti, L.; et al. DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin. Epigenet. 2015, 7, 61. [Google Scholar] [CrossRef]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary HCO(3)(−) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Spirli, C.; Nathanson, M.H.; Fiorotto, R.; Duner, E.; Denson, L.A.; Sanz, J.M.; Di Virgilio, F.; Okolicsanyi, L.; Casagrande, F.; Strazzabosco, M. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 2001, 121, 156–169. [Google Scholar] [CrossRef]
- Chang, J.C.; Go, S.; de Waart, D.R.; Munoz-Garrido, P.; Beuers, U.; Paulusma, C.C.; Oude Elferink, R. Soluble Adenylyl Cyclase Regulates Bile Salt-Induced Apoptosis in Human Cholangiocytes. Hepatology 2016, 64, 522–534. [Google Scholar] [CrossRef]
- Hohenester, S.; Wenniger, L.M.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Elferink, R.P.; Beuers, U. A biliary HCO3− umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver Int. 2013, 33, 312–320. [Google Scholar] [CrossRef]
- Chang, J.C.; Beuers, U.; Oude Elferink, R.P. The Emerging Role of Soluble Adenylyl Cyclase in Primary Biliary Cholangitis. Dig. Dis. 2017, 35, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Nakanuma, Y. Bile Acids and Deregulated Cholangiocyte Autophagy in Primary Biliary Cholangitis. Dig. Dis. 2017, 35, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Yoshimura-Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J. Gastroenterol. 2015, 50, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. An impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy in primary biliary cholangitis. Lab. Investig. 2018, 98, 745–754. [Google Scholar] [CrossRef]
- Hisamoto, S.; Shimoda, S.; Harada, K.; Iwasaka, S.; Onohara, S.; Chong, Y.; Nakamura, M.; Bekki, Y.; Yoshizumi, T.; Ikegami, T.; et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J. Autoimmun. 2016, 75, 150–160. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab. Investig. 2010, 90, 835–843. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J. Hepatol. 2010, 53, 318–325. [Google Scholar] [CrossRef]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Chemokine-chemokine receptor CCL2-CCR2 and CX3CL1-CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig. Dis. Sci. 2014, 59, 358–364. [Google Scholar] [CrossRef]
- Selmi, C.; Bowlus, C.L.; Gershwin, M.E.; Coppel, R.L. Primary biliary cirrhosis. Lancet 2011, 377, 1600–1609. [Google Scholar] [CrossRef]
- Bogdanos, D.P.; Pares, A.; Baum, H.; Caballeria, L.; Rigopoulou, E.I.; Ma, Y.; Burroughs, A.K.; Rodes, J.; Vergani, D. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J. Autoimmun. 2004, 22, 353–362. [Google Scholar] [CrossRef]
- Arbour, L.; Rupps, R.; Field, L.; Ross, P.; Erikson, A.; Henderson, H.; Hill, W.; Yoshida, E. Characteristics of primary biliary cirrhosis in British Columbia’s First Nations population. Can. J. Gastroenterol. 2005, 19, 305–310. [Google Scholar] [CrossRef]
- Selmi, C.; Gershwin, M.E. Chronic Autoimmune Epithelitis in Sjogren’s Syndrome and Primary Biliary Cholangitis: A Comprehensive Review. Rheumatol. Ther. 2017, 4, 263–279. [Google Scholar] [CrossRef]
- Lleo, A.; Maroni, L.; Glaser, S.; Alpini, G.; Marzioni, M. Role of cholangiocytes in primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 273–284. [Google Scholar] [CrossRef]
- Lleo, A.; Leung, P.S.C.; Hirschfield, G.M.; Gershwin, E.M. The Pathogenesis of Primary Biliary Cholangitis: A Comprehensive Review. Semin. Liver Dis. 2020, 40, 34–48. [Google Scholar] [CrossRef]
- Dahlqvist, G.; Gaouar, F.; Carrat, F.; Meurisse, S.; Chazouilleres, O.; Poupon, R.; Johanet, C.; Corpechot, C.; French Network of Immunology Laboratories. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology 2017, 65, 152–163. [Google Scholar] [CrossRef]
- Yamagiwa, S.; Kamimura, H.; Takamura, M.; Aoyagi, Y. Autoantibodies in primary biliary cirrhosis: Recent progress in research on the pathogenetic and clinical significance. World J. Gastroenterol. 2014, 20, 2606–2612. [Google Scholar] [CrossRef]
- Joshi, S.; Cauch-Dudek, K.; Heathcote, E.J.; Lindor, K.; Jorgensen, R.; Klein, R. Antimitochondrial antibody profiles: Are they valid prognostic indicators in primary biliary cirrhosis? Am. J. Gastroenterol. 2002, 97, 999–1002. [Google Scholar] [CrossRef]
- Tang, L.; Zhong, R.; He, X.; Wang, W.; Liu, J.; Zhu, Y.; Li, Y.; Hou, J. Evidence for the association between IgG-antimitochondrial antibody and biochemical response to ursodeoxycholic acid treatment in primary biliary cholangitis. J. Gastroenterol. Hepatol. 2017, 32, 659–666. [Google Scholar] [CrossRef]
- Zandanell, S.; Strasser, M.; Feldman, A.; Strebinger, G.; Aigner, G.; Niederseer, D.; Laimer, M.; Mussnig, B.; Paulweber, B.; Datz, C.; et al. Similar clinical outcome of AMA immunoblot-M2-negative compared to immunoblot-positive subjects over six years of follow-up. Postgrad. Med. 2021, 133, 291–298. [Google Scholar] [CrossRef]
- Muratori, P.; Muratori, L.; Gershwin, M.E.; Czaja, A.J.; Pappas, G.; MacCariello, S.; Granito, A.; Cassani, F.; Loria, P.; Lenzi, M.; et al. ‘True’ antimitochondrial antibody-negative primary biliary cirrhosis, low sensitivity of the routine assays, or both? Clin. Exp. Immunol. 2004, 135, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Yang, W.H.; Muratori, L.; Lim, M.J.; Nakajima, A.; Ferri, S.; Pappas, G.; Quarneti, C.; Bianchi, F.B.; Bloch, D.B.; et al. PML nuclear body component Sp140 is a novel autoantigen in primary biliary cirrhosis. Am. J. Gastroenterol. 2010, 105, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu. Rev. Pathol. 2013, 8, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Lian, Z.X.; Van de Water, J.; He, X.S.; Matsumura, S.; Kaplan, M.; Luketic, V.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J. Exp. Med. 2002, 195, 113–123. [Google Scholar] [CrossRef]
- Lleo, A.; Shimoda, S.; Ishibashi, H.; Gershwin, M.E. Primary biliary cirrhosis and autoimmune hepatitis: Apotopes and epitopes. J. Gastroenterol. 2011, 46 (Suppl. S1), 29–38. [Google Scholar] [CrossRef]
- Lleo, A.; Bowlus, C.L.; Yang, G.X.; Invernizzi, P.; Podda, M.; Van de Water, J.; Ansari, A.A.; Coppel, R.L.; Worman, H.J.; Gores, G.J.; et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 2010, 52, 987–998. [Google Scholar] [CrossRef]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Mackay, I.R.; Gores, G.J.; Ansari, A.A.; Van de Water, J.; Gershwin, M.E. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009, 49, 871–879. [Google Scholar] [CrossRef]
- Schrumpf, E.; Tan, C.; Karlsen, T.H.; Sponheim, J.; Bjorkstrom, N.K.; Sundnes, O.; Alfsnes, K.; Kaser, A.; Jefferson, D.M.; Ueno, Y.; et al. The biliary epithelium presents antigens to and activates natural killer T cells. Hepatology 2015, 62, 1249–1259. [Google Scholar] [CrossRef]
- Shimoda, S.; Harada, K.; Niiro, H.; Yoshizumi, T.; Soejima, Y.; Taketomi, A.; Maehara, Y.; Tsuneyama, K.; Nakamura, M.; Komori, A.; et al. Biliary epithelial cells and primary biliary cirrhosis: The role of liver-infiltrating mononuclear cells. Hepatology 2008, 47, 958–965. [Google Scholar] [CrossRef]
- Kita, H.; Matsumura, S.; He, X.S.; Ansari, A.A.; Lian, Z.X.; Van de Water, J.; Coppel, R.L.; Kaplan, M.M.; Gershwin, M.E. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J. Clin. Investig. 2002, 109, 1231–1240. [Google Scholar] [CrossRef]
- Yan, J.; Harvey, B.P.; Gee, R.J.; Shlomchik, M.J.; Mamula, M.J. B cells drive early T cell autoimmunity in vivo prior to dendritic cell-mediated autoantigen presentation. J. Immunol. 2006, 177, 4481–4487. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Tangye, S.G.; Moser, B.; Mackay, C.R. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 2005, 5, 853–865. [Google Scholar] [CrossRef]
- Yang, C.Y.; Ma, X.; Tsuneyama, K.; Huang, S.; Takahashi, T.; Chalasani, N.P.; Bowlus, C.L.; Yang, G.X.; Leung, P.S.; Ansari, A.A.; et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: Implications for therapy. Hepatology 2014, 59, 1944–1953. [Google Scholar] [CrossRef]
- Qian, C.; Jiang, T.; Zhang, W.; Ren, C.; Wang, Q.; Qin, Q.; Chen, J.; Deng, A.; Zhong, R. Increased IL-23 and IL-17 expression by peripheral blood cells of patients with primary biliary cirrhosis. Cytokine 2013, 64, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef]
- Bottcher, K.; Rombouts, K.; Saffioti, F.; Roccarina, D.; Rosselli, M.; Hall, A.; Luong, T.; Tsochatzis, E.A.; Thorburn, D.; Pinzani, M. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology 2018, 68, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lian, M.; Li, Y.; Zhang, W.; Wang, Q.; Wei, Y.; Zhang, J.; Chen, W.; Xiao, X.; Miao, Q.; et al. The immunobiology of mucosal-associated invariant T cell (MAIT) function in primary biliary cholangitis: Regulation by cholic acid-induced Interleukin-7. J. Autoimmun. 2018, 90, 64–75. [Google Scholar] [CrossRef]
- Setsu, T.; Yamagiwa, S.; Tominaga, K.; Kimura, N.; Honda, H.; Kamimura, H.; Tsuchiya, A.; Takamura, M.; Terai, S. Persistent reduction of mucosal-associated invariant T cells in primary biliary cholangitis. J. Gastroenterol. Hepatol. 2018, 33, 1286–1294. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Brussow, H. Microbiota and the human nature: Know thyself. Environ. Microbiol. 2015, 17, 10–15. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The microbiota in cirrhosis and its role in hepatic decompensation. J. Hepatol. 2021, 75 (Suppl. S1), S67–S81. [Google Scholar] [CrossRef] [PubMed]
- Liu Chen Kiow, J.; Vincent, C.; Sidani, S.; Bouin, M. High occurrence of small intestinal bacterial overgrowth in primary biliary cholangitis. Neurogastroenterol. Motil. 2019, 31, e13691. [Google Scholar] [CrossRef]
- Dyson, J.K.; Hirschfield, G.M.; Adams, D.H.; Beuers, U.; Mann, D.A.; Lindor, K.D.; Jones, D.E. Novel therapeutic targets in primary biliary cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 147–158. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Shimoda, S.; Harada, K.; Niiro, H.; Taketomi, A.; Maehara, Y.; Tsuneyama, K.; Kikuchi, K.; Nakanuma, Y.; Mackay, I.R.; Gershwin, M.E.; et al. CX3CL1 (fractalkine): A signpost for biliary inflammation in primary biliary cirrhosis. Hepatology 2010, 51, 567–575. [Google Scholar] [CrossRef]
- Harada, K.; Isse, K.; Nakanuma, Y. Interferon γ accelerates NF-κB activation of biliary epithelial cells induced by Toll-like receptor and ligand interaction. J. Clin. Pathol. 2006, 59, 184–190. [Google Scholar] [CrossRef]
- Atif, M.; Warner, S.; Oo, Y.H. Linking the gut and liver: Crosstalk between regulatory T cells and mucosa-associated invariant T cells. Hepatol. Int. 2018, 12, 305–314. [Google Scholar] [CrossRef]
- Jeffery, H.C.; van Wilgenburg, B.; Kurioka, A.; Parekh, K.; Stirling, K.; Roberts, S.; Dutton, E.E.; Hunter, S.; Geh, D.; Braitch, M.K.; et al. Biliary epithelium and liver B cells exposed to bacteria activate intrahepatic MAIT cells through MR1. J. Hepatol. 2016, 64, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef]
- Inamine, T.; Schnabl, B. Immunoglobulin A and liver diseases. J. Gastroenterol. 2018, 53, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Baragiotta, A.; Pizzuti, D.; Martines, D.; Cecchetto, A.; Chiarelli, S. Mucosal IgA defect in primary biliary cirrhosis. Am. J. Gastroenterol. 2002, 97, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Chazouilleres, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Campisi, L.; Barbet, G.; Ding, Y.; Esplugues, E.; Flavell, R.A.; Blander, J.M. Apoptosis in response to microbial infection induces autoreactive TH17 cells. Nat. Immunol. 2016, 17, 1084–1092. [Google Scholar] [CrossRef]
- Haruta, I.; Kikuchi, K.; Hashimoto, E.; Nakamura, M.; Miyakawa, H.; Hirota, K.; Shibata, N.; Kato, H.; Arimura, Y.; Kato, Y.; et al. Long-term bacterial exposure can trigger nonsuppurative destructive cholangitis associated with multifocal epithelial inflammation. Lab. Investig. 2010, 90, 577–588. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Lemoinne, S.; Marteau, P. Gut microbial profile in primary biliary cholangitis: Towards bioindicators. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 507–508. [Google Scholar] [CrossRef]
- Lv, L.X.; Fang, D.Q.; Shi, D.; Chen, D.Y.; Yan, R.; Zhu, Y.X.; Chen, Y.F.; Shao, L.; Guo, F.F.; Wu, W.R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The Diagnosis and Management of Patients with Primary Biliary Cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J. Hepatol. 2001, 35, 134–146. [Google Scholar] [CrossRef]
- Hofmann, A.F. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand. J. Gastroenterol. 1994, 29 (Suppl. S204), 1–15. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Graziadei, I.W. Review article: Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment. Pharmacol. Ther. 1999, 13, 979–996. [Google Scholar] [CrossRef]
- You, H.; Ma, X.; Efe, C.; Wang, G.; Jeong, S.H.; Abe, K.; Duan, W.; Chen, S.; Kong, Y.; Zhang, D.; et al. APASL clinical practice guidance: The diagnosis and management of patients with primary biliary cholangitis. Hepatol. Int. 2022, 16, 1–23. [Google Scholar] [CrossRef]
- Carbone, M.; Nardi, A.; Flack, S.; Carpino, G.; Varvaropoulou, N.; Gavrila, C.; Spicer, A.; Badrock, J.; Bernuzzi, F.; Cardinale, V.; et al. Pretreatment prediction of response to ursodeoxycholic acid in primary biliary cholangitis: Development and validation of the UDCA Response Score. Lancet Gastroenterol. Hepatol. 2018, 3, 626–634. [Google Scholar] [CrossRef]
- Lammers, W.J.; van Buuren, H.R.; Hirschfield, G.M.; Janssen, H.L.; Invernizzi, P.; Mason, A.L.; Ponsioen, C.Y.; Floreani, A.; Corpechot, C.; Mayo, M.J.; et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: An international follow-up study. Gastroenterology 2014, 147, 1338–1349.e5. [Google Scholar] [CrossRef]
- Pares, A.; Caballeria, L.; Rodes, J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic Acid. Gastroenterology 2006, 130, 715–720. [Google Scholar] [CrossRef]
- Angulo, P.; Lindor, K.D.; Therneau, T.M.; Jorgensen, R.A.; Malinchoc, M.; Kamath, P.S.; Dickson, E.R. Utilization of the Mayo risk score in patients with primary biliary cirrhosis receiving ursodeoxycholic acid. Liver 1999, 19, 115–121. [Google Scholar] [CrossRef]
- Corpechot, C.; Abenavoli, L.; Rabahi, N.; Chretien, Y.; Andreani, T.; Johanet, C.; Chazouilleres, O.; Poupon, R. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008, 48, 871–877. [Google Scholar] [CrossRef]
- Kuiper, E.M.; Hansen, B.E.; de Vries, R.A.; den Ouden-Muller, J.W.; van Ditzhuijsen, T.J.; Haagsma, E.B.; Houben, M.H.; Witteman, B.J.; van Erpecum, K.J.; van Buuren, H.R.; et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology 2009, 136, 1281–1287. [Google Scholar] [CrossRef]
- Azemoto, N.; Abe, M.; Murata, Y.; Hiasa, Y.; Hamada, M.; Matsuura, B.; Onji, M. Early biochemical response to ursodeoxycholic acid predicts symptom development in patients with asymptomatic primary biliary cirrhosis. J. Gastroenterol. 2009, 44, 630–634. [Google Scholar] [CrossRef]
- Kumagi, T.; Guindi, M.; Fischer, S.E.; Arenovich, T.; Abdalian, R.; Coltescu, C.; Heathcote, E.J.; Hirschfield, G.M. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am. J. Gastroenterol. 2010, 105, 2186–2194. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouilleres, O.; Poupon, R. Early primary biliary cirrhosis: Biochemical response to treatment and prediction of long-term outcome. J. Hepatol. 2011, 55, 1361–1367. [Google Scholar] [CrossRef]
- Sepe, V.; Distrutti, E.; Fiorucci, S.; Zampella, A. Farnesoid X receptor modulators 2014–present: A patent review. Expert Opin. Ther. Pat. 2018, 28, 351–364. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef]
- Triantis, V.; Saeland, E.; Bijl, N.; Oude-Elferink, R.P.; Jansen, P.L. Glycosylation of fibroblast growth factor receptor 4 is a key regulator of fibroblast growth factor 19-mediated down-regulation of cytochrome P450 7A1. Hepatology 2010, 52, 656–666. [Google Scholar] [CrossRef]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Trauner, M.; Nevens, F.; Shiffman, M.L.; Drenth, J.P.H.; Bowlus, C.L.; Vargas, V.; Andreone, P.; Hirschfield, G.M.; Pencek, R.; Malecha, E.S.; et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol. Hepatol. 2019, 4, 445–453. [Google Scholar] [CrossRef]
- Hegade, V.S.; Kendrick, S.F.; Dobbins, R.L.; Miller, S.R.; Thompson, D.; Richards, D.; Storey, J.; Dukes, G.E.; Corrigan, M.; Oude Elferink, R.P.; et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: A double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 2017, 389, 1114–1123. [Google Scholar] [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Regulation of bile acid and cholesterol metabolism by PPARs. PPAR Res. 2009, 2009, 501739. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Xia, X.; Jung, D.; Webb, P.; Zhang, A.; Zhang, B.; Li, L.; Ayers, S.D.; Gabbi, C.; Ueno, Y.; Gustafsson, J.A.; et al. Liver X receptor beta and peroxisome proliferator-activated receptor delta regulate cholesterol transport in murine cholangiocytes. Hepatology 2012, 56, 2288–2296. [Google Scholar] [CrossRef]
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U.; et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 2009, 15, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Isse, K.; Kamihira, T.; Shimoda, S.; Nakanuma, Y. Th1 cytokine-induced downregulation of PPARγ in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology 2005, 41, 1329–1338. [Google Scholar] [CrossRef]
- Nozaki, Y.; Harada, K.; Sanzen, T.; Nakanuma, Y. PPARγ ligand attenuates portal inflammation in the MRL-lpr mouse: A new strategy to restrain cholangiopathy in primary biliary cirrhosis. Med. Mol. Morphol. 2013, 46, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Han, X.F.; Wang, Q.X.; Liu, Y.; You, Z.R.; Bian, Z.L.; Qiu, D.K.; Ma, X. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J. Dig. Dis. 2012, 13, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Sato, Y.; Ueno, T.; Sata, M. Fenofibrate treatment in patients with primary biliary cirrhosis. Am. J. Gastroenterol. 2002, 97, 2147–2149. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Ou, X.; Wang, X.; Wang, Y.; Zhao, X.; Wang, Q.; Wu, X.; Zhang, W.; Ma, H.; You, H.; et al. Efficacy and safety of fenofibrate add-on therapy for patients with primary biliary cholangitis and a suboptimal response to UDCA. Rev. Esp. Enferm. Dig. 2018, 110, 557–563. [Google Scholar] [CrossRef]
- Yin, Q.; Li, J.; Xia, Y.; Zhang, R.; Wang, J.; Lu, W.; Zhou, Y.; Zheng, Y.; Abudumijiti, H.; Chen, R.; et al. Systematic review and meta-analysis: Bezafibrate in patients with primary biliary cirrhosis. Drug Des. Dev. Ther. 2015, 9, 5407–5419. [Google Scholar] [CrossRef]
- Reig, A.; Sese, P.; Pares, A. Effects of Bezafibrate on Outcome and Pruritus in Primary Biliary Cholangitis with Suboptimal Ursodeoxycholic Acid Response. Am. J. Gastroenterol. 2018, 113, 49–55. [Google Scholar] [CrossRef]
- Honda, A.; Tanaka, A.; Kaneko, T.; Komori, A.; Abe, M.; Inao, M.; Namisaki, T.; Hashimoto, N.; Kawata, K.; Takahashi, A.; et al. Bezafibrate Improves GLOBE and UK-PBC Scores and Long-Term Outcomes in Patients with Primary Biliary Cholangitis. Hepatology 2019, 70, 2035–2046. [Google Scholar] [CrossRef]
- Bolier, R.; de Vries, E.S.; Pares, A.; Helder, J.; Kemper, E.M.; Zwinderman, K.; Elferink, R.P.O.; Beuers, U.; Netherlands Association for the Study of the Liver Cholestatic Liver Diseases Study Group. Fibrates for the treatment of cholestatic itch (FITCH): Study protocol for a randomized controlled trial. Trials 2017, 18, 230. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Caldwell, S.H.; Pyrsopoulos, N. Results of a Phase 2, Prospective, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Safety, Tolerability, and Efficacy of Saroglitazar Magnesium in Patients with Primary Biliary Cholangitis (EPICS). Gastroenterol. Hepatol. 2021, 17, 8. [Google Scholar]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Kowdley, K.V.; Shiffman, M.L. ENHANCE: Safety and Efficacy of Seladelpar in Patients with Primary Biliary Cholangitis-A Phase 3, International, Randomized, Placebo-Controlled Study. Gastroenterol. Hepatol. 2021, 17, 5–6. [Google Scholar]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; Levy, C.; Bowlus, C.L.; Jones, D.E.; Steinberg, A.; McWherter, C.A.; Choi, Y.J. Seladelpar improved measures of pruritus, sleep, and fatigue and decreased serum bile acids in patients with primary biliary cholangitis. Liver Int. 2022, 42, 112–123. [Google Scholar] [CrossRef]
- Rautiainen, H.; Karkkainen, P.; Karvonen, A.L.; Nurmi, H.; Pikkarainen, P.; Nuutinen, H.; Farkkila, M. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: A three-year randomized trial. Hepatology 2005, 41, 747–752. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Kupcinskas, L.; Ott, P.; Bergquist, A.; Farkkila, M.; Manns, M.P.; Pares, A.; Spengler, U.; Stiess, M.; et al. A placebo-controlled randomised trial of budesonide for PBC following an insufficient response to UDCA. J. Hepatol. 2021, 74, 321–329. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Di Tullio, G.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012, 142, 355–365.e4. [Google Scholar] [CrossRef]
- Luo, J.; Ko, B.; Elliott, M.; Zhou, M.; Lindhout, D.A.; Phung, V.; To, C.; Learned, R.M.; Tian, H.; DePaoli, A.M.; et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci. Transl. Med. 2014, 6, 247ra100. [Google Scholar] [CrossRef]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.P.; et al. A mouse model of hepatocellular carcinoma: Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients with Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Gege, C.; Hambruch, E.; Hambruch, N.; Kinzel, O.; Kremoser, C. Nonsteroidal FXR Ligands: Current Status and Clinical Applications. Handb. Exp. Pharmacol. 2019, 256, 167–205. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wei, G.; Huang, P.; Li, W.; Qi, X.; Lin, Y.; Vaid, K.A.; Wang, J.; Zhang, S.; Li, Y.; et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020, 40, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Mogul, A.; Corsi, K.; McAuliffe, L. Baricitinib: The Second FDA-Approved JAK Inhibitor for the Treatment of Rheumatoid Arthritis. Ann. Pharmacother. 2019, 53, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Asuri, S.; McIntosh, S.; Taylor, V.; Rokeby, A.; Kelly, J.; Shumansky, K.; Field, L.L.; Yoshida, E.M.; Arbour, L. Primary Biliary Cholangitis in British Columbia First Nations: Clinical Features and Discovery of Novel Genetic Susceptibility Loci. Liver Int. 2018, 38, 940–948. [Google Scholar] [CrossRef]
- Gordon, S.C.; Trudeau, S.; Regev, A.; Uhas, J.M.; Chakladar, S.; Pinto-Correia, A.; Gottlieb, K.; Schlichting, D. Baricitinib and primary biliary cholangitis. J. Transl. Autoimmun. 2021, 4, 100107. [Google Scholar] [CrossRef]
- Andringa, K.K.; King, A.L.; Eccleston, H.B.; Mantena, S.K.; Landar, A.; Jhala, N.C.; Dickinson, D.A.; Squadrito, G.L.; Bailey, S.M. Analysis of the liver mitochondrial proteome in response to ethanol and S-adenosylmethionine treatments: Novel molecular targets of disease and hepatoprotection. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G732–G745. [Google Scholar] [CrossRef]
- Caballero, F.; Fernandez, A.; Matias, N.; Martinez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef]
- Ko, K.; Yang, H.; Noureddin, M.; Iglesia-Ara, A.; Xia, M.; Wagner, C.; Luka, Z.; Mato, J.M.; Lu, S.C. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab. Investig. 2008, 88, 1121–1129. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Mingorance, J.; Duran, C.; Pajares, M.A. S-adenosyl-L-methionine synthetase and methionine metabolism deficiencies in cirrhosis. Adv. Exp. Med. Biol. 1994, 368, 113–117. [Google Scholar] [CrossRef]
- Wunsch, E.; Raszeja-Wyszomirska, J.; Barbier, O.; Milkiewicz, M.; Krawczyk, M.; Milkiewicz, P. Effect of S-adenosyl-L-methionine on liver biochemistry and quality of life in patients with primary biliary cholangitis treated with ursodeoxycholic acid. A prospective, open label pilot study. J. Gastrointestin. Liver Dis. 2018, 27, 273–279. [Google Scholar] [CrossRef]
- Kilanczyk, E.; Banales, J.M.; Wunsch, E.; Barbier, O.; Avila, M.A.; Mato, J.M.; Milkiewicz, M.; Milkiewicz, P. S-adenosyl-L-methionine (SAMe) halts the autoimmune response in patients with primary biliary cholangitis (PBC) via antioxidant and S-glutathionylation processes in cholangiocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165895. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, K.; Li, F.; Gu, Z.; Liu, Q.; He, L.; Shao, T.; Song, Q.; Zhu, F.; Zhang, L.; et al. Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice. Hepatology 2020, 71, 2050–2066. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Ljujic, B.; Stojkovic, P.; Lukic, A.; Arsenijevic, N.; Stojkovic, M. Human stem cell research and regenerative medicine--present and future. Br. Med. Bull. 2011, 99, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Liu, H.; Li, Y.; Fu, J.; Sun, Y.; Xu, R.; Lin, H.; Wang, S.; Lv, S.; et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S1), 85–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, Q.; Chen, H.; Wang, K.; Shan, G.L.; Kong, F.; Yang, Y.J.; Li, Y.Z.; Zhang, X.; Dong, F.; et al. Allogeneic bone marrow mesenchymal stem cell transplantation in patients with UDCA-resistant primary biliary cirrhosis. Stem Cells Dev. 2014, 23, 2482–2489. [Google Scholar] [CrossRef]
- Isse, K.; Harada, K.; Zen, Y.; Kamihira, T.; Shimoda, S.; Harada, M.; Nakanuma, Y. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology 2005, 41, 506–516. [Google Scholar] [CrossRef]
- Tabuchi, H.; Katsurabara, T.; Mori, M.; Aoyama, M.; Obara, T.; Yasuda, N.; Kawano, T.; Imai, T.; Ieiri, I.; Kumagai, Y. Pharmacokinetics, Pharmacodynamics, and Safety of E6011, a Novel Humanized Antifractalkine (CX3CL1) Monoclonal Antibody: A Randomized, Double-Blind, Placebo-Controlled Single-Ascending-Dose Study. J. Clin. Pharmacol. 2019, 59, 688–701. [Google Scholar] [CrossRef]
- Chapman, M.H.; Thorburn, D.; Hirschfield, G.M.; Webster, G.G.J.; Rushbrook, S.M.; Alexander, G.; Collier, J.; Dyson, J.K.; Jones, D.E.; Patanwala, I.; et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 2019, 68, 1356–1378. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 2501–2502. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Sirpal, S.; Chandok, N. Primary sclerosing cholangitis: Diagnostic and management challenges. Clin. Exp. Gastroenterol. 2017, 10, 265–273. [Google Scholar] [CrossRef]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Jiang, X.; Karlsen, T.H. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, A.; Montgomery, S.M.; Bahmanyar, S.; Olsson, R.; Danielsson, A.; Lindgren, S.; Prytz, H.; Hultcrantz, R.; Loof, L.A.; Sandberg-Gertzen, H.; et al. Increased risk of primary sclerosing cholangitis and ulcerative colitis in first-degree relatives of patients with primary sclerosing cholangitis. Clin. Gastroenterol. Hepatol. 2008, 6, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.G.; Juran, B.D.; Mucha, S.; Folseraas, T.; Jostins, L.; Melum, E.; Kumasaka, N.; Atkinson, E.J.; Schlicht, E.M.; Liu, J.Z.; et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 2017, 49, 269–273. [Google Scholar] [CrossRef]
- Ataseven, H.; Parlak, E.; Yuksel, I.; Basar, O.; Ertugrul, I.; Sasmaz, N.; Sahin, B. Primary sclerosing cholangitis in Turkish patients: Characteristic features and prognosis. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 312–315. [Google Scholar]
- Tanaka, A.; Tazuma, S.; Okazaki, K.; Tsubouchi, H.; Inui, K.; Takikawa, H. Nationwide survey for primary sclerosing cholangitis and IgG4-related sclerosing cholangitis in Japan. J. Hepatobiliary Pancreat. Sci. 2014, 21, 43–50. [Google Scholar] [CrossRef]
- Saarinen, S.; Olerup, O.; Broome, U. Increased frequency of autoimmune diseases in patients with primary sclerosing cholangitis. Am. J. Gastroenterol. 2000, 95, 3195–3199. [Google Scholar] [CrossRef]
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismuller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675. [Google Scholar] [CrossRef]
- Hov, J.R.; Kosmoliaptsis, V.; Traherne, J.A.; Olsson, M.; Boberg, K.M.; Bergquist, A.; Schrumpf, E.; Bradley, J.A.; Taylor, C.J.; Lie, B.A.; et al. Electrostatic modifications of the human leukocyte antigen-DR P9 peptide-binding pocket and susceptibility to primary sclerosing cholangitis. Hepatology 2011, 53, 1967–1976. [Google Scholar] [CrossRef]
- Hov, J.R.; Lleo, A.; Selmi, C.; Woldseth, B.; Fabris, L.; Strazzabosco, M.; Karlsen, T.H.; Invernizzi, P. Genetic associations in Italian primary sclerosing cholangitis: Heterogeneity across Europe defines a critical role for HLA-C. J. Hepatol. 2010, 52, 712–717. [Google Scholar] [CrossRef]
- Eksteen, B. Advances and controversies in the pathogenesis and management of primary sclerosing cholangitis. Br. Med. Bull. 2014, 110, 89–98. [Google Scholar] [CrossRef][Green Version]
- Hofmann, A.F. Bile acids: Trying to understand their chemistry and biology with the hope of helping patients. Hepatology 2009, 49, 1403–1418. [Google Scholar] [CrossRef]
- Hohenester, S.; Gates, A.; Wimmer, R.; Beuers, U.; Anwer, M.S.; Rust, C.; Webster, C.R. Phosphatidylinositol-3-kinase p110gamma contributes to bile salt-induced apoptosis in primary rat hepatocytes and human hepatoma cells. J. Hepatol. 2010, 53, 918–926. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Bell, L.N.; Wulff, J.; Comerford, M.; Vuppalanchi, R.; Chalasani, N. Serum metabolic signatures of primary biliary cirrhosis and primary sclerosing cholangitis. Liver Int. 2015, 35, 263–274. [Google Scholar] [CrossRef]
- Deutschmann, K.; Reich, M.; Klindt, C.; Droge, C.; Spomer, L.; Haussinger, D.; Keitel, V. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1319–1325. [Google Scholar] [CrossRef]
- Zhang, J.H.; Nolan, J.D.; Kennie, S.L.; Johnston, I.M.; Dew, T.; Dixon, P.H.; Williamson, C.; Walters, J.R. Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G940–G948. [Google Scholar] [CrossRef]
- Milkiewicz, M.; Klak, M.; Kempinska-Podhorodecka, A.; Wiechowska-Kozlowska, A.; Urasinska, E.; Blatkiewicz, M.; Wunsch, E.; Elias, E.; Milkiewicz, P. Impaired Hepatic Adaptation to Chronic Cholestasis induced by Primary Sclerosing Cholangitis. Sci. Rep. 2016, 6, 39573. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Gigliozzi, A.; Attili, A.F. Regulation and deregulation of cholangiocyte proliferation. J. Hepatol. 2000, 33, 333–340. [Google Scholar] [CrossRef]
- Carpino, G.; Cardinale, V.; Renzi, A.; Hov, J.R.; Berloco, P.B.; Rossi, M.; Karlsen, T.H.; Alvaro, D.; Gaudio, E. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 2015, 63, 1220–1228. [Google Scholar] [CrossRef]
- Karlsen, T.H. Primary sclerosing cholangitis: 50 years of a gut-liver relationship and still no love? Gut 2016, 65, 1579–1581. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.R.; Legrand, N.; Papiernik, M.; Freitas, A.A. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 2002, 169, 4850–4860. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Cadoret, A.; Rautou, P.E.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef]
- Ferreira-Gonzalez, S.; Lu, W.Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef]
- Kyritsi, K.; Francis, H.; Zhou, T.; Ceci, L.; Wu, N.; Yang, Z.; Meng, F.; Chen, L.; Baiocchi, L.; Kundu, D.; et al. Downregulation of p16 Decreases Biliary Damage and Liver Fibrosis in the Mdr2(/) Mouse Model of Primary Sclerosing Cholangitis. Gene Expr. 2020, 20, 89–103. [Google Scholar] [CrossRef]
- Trussoni, C.E.; O’Hara, S.P.; LaRusso, N.F. Cellular senescence in the cholangiopathies: A driver of immunopathology and a novel therapeutic target. Semin. Immunopathol. 2022. [Google Scholar] [CrossRef]
- Cameron, R.G.; Blendis, L.M.; Neuman, M.G. Accumulation of macrophages in primary sclerosing cholangitis. Clin. Biochem. 2001, 34, 195–201. [Google Scholar] [CrossRef]
- Grant, A.J.; Lalor, P.F.; Hubscher, S.G.; Briskin, M.; Adams, D.H. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology 2001, 33, 1065–1072. [Google Scholar] [CrossRef]
- Eksteen, B.; Grant, A.J.; Miles, A.; Curbishley, S.M.; Lalor, P.F.; Hubscher, S.G.; Briskin, M.; Salmon, M.; Adams, D.H. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J. Exp. Med. 2004, 200, 1511–1517. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Gut-liver immunity. J. Hepatol. 2016, 64, 1187–1189. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Mucosal immunity in liver autoimmunity: A comprehensive review. J. Autoimmun. 2013, 46, 97–111. [Google Scholar] [CrossRef]
- Worthington, J.; Cullen, S.; Chapman, R. Immunopathogenesis of primary sclerosing cholangitis. Clin. Rev. Allergy Immunol. 2005, 28, 93–103. [Google Scholar] [CrossRef]
- Ruhlemann, M.; Liwinski, T.; Heinsen, F.A.; Bang, C.; Zenouzi, R.; Kummen, M.; Thingholm, L.; Tempel, M.; Lieb, W.; Karlsen, T.; et al. Consistent alterations in faecal microbiomes of patients with primary sclerosing cholangitis independent of associated colitis. Aliment. Pharmacol. Ther. 2019, 50, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygard, S.; Vesterhus, M.; Hoivik, M.L.; Troseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of Antibiotic Therapy in Primary Sclerosing Cholangitis with and without Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutos, G.; Ghosh, S.; Rossiter, A.E.; Loman, N.; van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease with Bile Acid Pathways. J. Crohns Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef]
- Jiang, B.; Yuan, G.; Wu, J.; Wu, Q.; Li, L.; Jiang, P. Prevotella copri ameliorates cholestasis and liver fibrosis in primary sclerosing cholangitis by enhancing the FXR signalling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166320. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Chazouilleres, O.; Housset, C.; Sokol, H.; Saint-Antoine IBD Network. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef]
- Hong, K.H.; Kim, J.W.; Jang, S.J.; Yu, E.; Kim, E.C. Liver cirrhosis caused by Exophiala dermatitidis. J. Med. Microbiol. 2009, 58, 674–677. [Google Scholar] [CrossRef]
- Kremer, A.E.; Namer, B.; Bolier, R.; Fischer, M.J.; Oude Elferink, R.P.; Beuers, U. Pathogenesis and Management of Pruritus in PBC and PSC. Dig. Dis. 2015, 33 (Suppl. S2), 164–175. [Google Scholar] [CrossRef]
- De Vries, E.; Bolier, R.; Goet, J.; Pares, A.; Verbeek, J.; de Vree, M.; Drenth, J.; van Erpecum, K.; van Nieuwkerk, K.; van der Heide, F.; et al. Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial. Gastroenterology 2021, 160, 734–743.e6. [Google Scholar] [CrossRef] [PubMed]
- Kumada, H.; Miyakawa, H.; Muramatsu, T.; Ando, N.; Oh, T.; Takamori, K.; Nakamoto, H. Efficacy of nalfurafine hydrochloride in patients with chronic liver disease with refractory pruritus: A randomized, double-blind trial. Hepatol. Res. 2017, 47, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Dull, M.M.; Wolf, K.; Vetter, M.; Dietrich, P.; Neurath, M.F.; Kremer, A.E. Endogenous Opioid Levels Do Not Correlate with Itch Intensity and Therapeutic Interventions in Hepatic Pruritus. Front. Med. 2021, 8, 641163. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Harrison, M.E.; American College of Gastroenterology. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2015, 110, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Ebrahimi-Daryani, N.; Nouri-Taromlou, M.K.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointestin. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Tabibian, J.H.; Carey, E.J.; Gostout, C.J.; Lindor, K.D. Dominant strictures in primary sclerosing cholangitis: A multicenter survey of clinical definitions and practices. Hepatol. Commun. 2018, 2, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, A.; Rudolph, G.; Kloters-Plachky, P.; Sauer, P.; Walker, S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: Outcome after endoscopic treatment. J. Hepatol. 2002, 36, 151–156. [Google Scholar] [CrossRef]
- Bjornsson, E.; Lindqvist-Ottosson, J.; Asztely, M.; Olsson, R. Dominant strictures in patients with primary sclerosing cholangitis. Am. J. Gastroenterol. 2004, 99, 502–508. [Google Scholar] [CrossRef] [PubMed]
- European Society of Gastrointestinal Endoscopy; European Association for the Study of the Liver. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. J. Hepatol. 2017, 66, 1265–1281. [Google Scholar] [CrossRef]
- Rupp, C.; Hippchen, T.; Bruckner, T.; Kloters-Plachky, P.; Schaible, A.; Koschny, R.; Stiehl, A.; Gotthardt, D.N.; Sauer, P. Effect of scheduled endoscopic dilatation of dominant strictures on outcome in patients with primary sclerosing cholangitis. Gut 2019, 68, 2170–2178. [Google Scholar] [CrossRef]
- Ferreira, M.; Ribeiro, I.B.; de Moura, D.T.H.; McCarty, T.R.; da Ponte Neto, A.M.; Farias, G.F.A.; de Miranda Neto, A.A.; de Oliveira, P.; Bernardo, W.M.; de Moura, E.G.H. Stent versus Balloon Dilation for the Treatment of Dominant Strictures in Primary Sclerosing Cholangitis: A Systematic Review and Meta-Analysis. Clin. Endosc. 2021, 54, 833–842. [Google Scholar] [CrossRef]
- Song, J.; Li, Y.; Bowlus, C.L.; Yang, G.; Leung, P.S.C.; Gershwin, M.E. Cholangiocarcinoma in Patients with Primary Sclerosing Cholangitis (PSC): A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 58, 134–149. [Google Scholar] [CrossRef]
- Said, K.; Glaumann, H.; Bergquist, A. Gallbladder disease in patients with primary sclerosing cholangitis. J. Hepatol. 2008, 48, 598–605. [Google Scholar] [CrossRef]
- Mainprize, K.S.; Gould, S.W.; Gilbert, J.M. Surgical management of polypoid lesions of the gallbladder. Br. J. Surg. 2000, 87, 414–417. [Google Scholar] [CrossRef]
- Torabi Sagvand, B.; Edwards, K.; Shen, B. Frequency, Risk Factors, and Outcome of Gallbladder Polyps in Patients with Primary Sclerosing Cholangitis: A Case-Control Study. Hepatol. Commun. 2018, 2, 1440–1445. [Google Scholar] [CrossRef]
- Hildebrand, T.; Pannicke, N.; Dechene, A.; Gotthardt, D.N.; Kirchner, G.; Reiter, F.P.; Sterneck, M.; Herzer, K.; Lenzen, H.; Rupp, C.; et al. Biliary strictures and recurrence after liver transplantation for primary sclerosing cholangitis: A retrospective multicenter analysis. Liver Transpl. 2016, 22, 42–52. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Crothers, H.; Mytton, J.; Bosch, S.; Iqbal, T.; Ferguson, J.; Hirschfield, G.M. Effects of Primary Sclerosing Cholangitis on Risks of Cancer and Death in People with Inflammatory Bowel Disease, Based on Sex, Race, and Age. Gastroenterology 2020, 159, 915–928. [Google Scholar] [CrossRef]
- Harms, M.H.; van Buuren, H.R.; Corpechot, C.; Thorburn, D.; Janssen, H.L.A.; Lindor, K.D.; Hirschfield, G.M.; Pares, A.; Floreani, A.; Mayo, M.J.; et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2019, 71, 357–365. [Google Scholar] [CrossRef]
- Wunsch, E.; Trottier, J.; Milkiewicz, M.; Raszeja-Wyszomirska, J.; Hirschfield, G.M.; Barbier, O.; Milkiewicz, P. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014, 60, 931–940. [Google Scholar] [CrossRef]
- Stokkeland, K.; Hoijer, J.; Bottai, M.; Soderberg-Lofdal, K.; Bergquist, A. Statin Use Is Associated with Improved Outcomes of Patients with Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2019, 17, 1860–1866.e1. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, J.; Chen, M.; Yang, H.; Learned, R.M.; DePaoli, A.M.; Tian, H.; Ling, L. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J. Hepatol. 2017, 66, 1182–1192. [Google Scholar] [CrossRef]
- Mizuno, S.; Hirano, K.; Isayama, H.; Watanabe, T.; Yamamoto, N.; Nakai, Y.; Sasahira, N.; Tada, M.; Omata, M.; Koike, K. Prospective study of bezafibrate for the treatment of primary sclerosing cholangitis. J. Hepatobiliary Pancreat. Sci. 2015, 22, 766–770. [Google Scholar] [CrossRef]
- Hirano, F.; Kobayashi, A.; Makino, I. Inhibition of TNF-alpha-induced RANTES expression in human hepatocyte-derived cells by fibrates, the hypolipidemic drugs. Int. Immunopharmacol. 2003, 3, 225–232. [Google Scholar] [CrossRef]
- Schierwagen, R.; Uschner, F.E.; Magdaleno, F.; Klein, S.; Trebicka, J. Rationale for the use of statins in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G407–G412. [Google Scholar] [CrossRef]
- Chung, B.K.; Hirschfield, G.M. Immunogenetics in primary sclerosing cholangitis. Curr. Opin. Gastroenterol. 2017, 33, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.H.; Damman, J.; Shah, S.B.; Davies, Y.; Hurwitz, M.; Stephen, M.; Lemos, L.M.; Carey, E.J.; Lindor, K.D.; Buness, C.W.; et al. Open-label prospective therapeutic clinical trials: Oral vancomycin in children and adults with primary sclerosing cholangitis. Scand. J. Gastroenterol. 2020, 55, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.U.; Altorjay, I.; Farkkila, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Carey, E.J.; Eaton, J.; Clayton, M.; Gossard, A.; Iqbal, S.; Ullah, H.; Zhang, N.; Butterfield, R.; Lindor, K.D. A pilot study of vidofludimus calcium for treatment of primary sclerosing cholangitis. Hepatol. Commun. 2022. [Google Scholar] [CrossRef]
- Eksteen, B.; Bowlus, C.L.; Montano-Loza, A.J.; Lefebvre, E.; Fischer, L.; Vig, P.; Martins, E.B.; Ahmad, J.; Yimam, K.K.; Pockros, P.J.; et al. Efficacy and Safety of Cenicriviroc in Patients with Primary Sclerosing Cholangitis: PERSEUS Study. Hepatol. Commun. 2021, 5, 478–490. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chazouilleres, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients with Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Gidwaney, N.G.; Pawa, S.; Das, K.M. Pathogenesis and clinical spectrum of primary sclerosing cholangitis. World J Gastroenterol. 2017, 23, 2459–2469. [Google Scholar] [CrossRef]


{kind=link}
| Criteria | Definition of Response (Time to Evaluation, Months) | Ref. |
|---|---|---|
| Barcelona | >40% decrease or normalization of ALP (12) | [109] |
| Mayo | ALP < 2 × ULN (6) | [110] |
| Paris I | ALP ≤ 3 × ULN and AST ≤ 2 × ULN and normalization of bilirubin (12) | [111] |
| Rotterdam | Normalization of bilirubin and/or albumin (12) | [112] |
| Ehime | ≥70% decrease or normalization of GGT (6) | [113] |
| Toronto | ALP ≤ 1.67 × ULN (24) | [114] |
| Paris II | ALP and AST ≤ 1.5 × ULN and normalization of bilirubin (12) | [115] |
| Agent | Mechanism | Clinical Trial Stage | Main Results | Ref. |
|---|---|---|---|---|
| Bezafibrate | Panspecific PPAR agonist | Phase III (BEZURSO trial) |
| [94] |
| Elafibranor | PPARα and PPARδ agonist | Phase II (NCT03124108) |
| [140] |
| Saroglitazar | PPARα and PPARγ agonist | Phase II (EPICS) |
| [141] |
| Tropifexor | FXR agonist | Phase II (NCT02516605) | Not yet published | |
| Cilofexor | FXR agonist | Phase II (NCT02943447) | Not yet published | |
| EDP-305 | FXR agonist | Phase II (NCT03394924) | Not yet published | |
| Baricitinib | JAK 1 and 2 inhibitor | Proof-of-concept study |
| [158] |
| S-adenosyl-L-methionine | 17-beta-estradiol glucuronide-induced cholestasis reversal agent | Pilot, open-label study |
| [163] |
| Probiotics | Regulation of bile acid homeostasis | Phase II (NCT03521297) | Data yet to be collected | |
| Mesenchymal Stem Cells | Immunoregulation | NCT03668145 | Data yet to be collected |
| Agent | Mechanism | Clinical Trial Stage | Main Results | Ref. |
|---|---|---|---|---|
| 24-norursodeoxycholic acid (norUDCA) | Side chain-shortened C23 homolog of UDCA | Phase II (NUC-3) Phase III (NCT03872921) |
| [248] |
| Berberine ursodeoxycholate (BUDCA) | Ionic salt of two active moieties, berberine and UDCA | Phase II |
| |
| Obeticholic acid | FXR agonist | Phase II (AESOP) |
| [249] |
| Cilofexor | FXR agonist | Phase III (PRIMIS) |
| |
| Vidofludimus calcium | FXR agonists + dihydroorotate dehydrogenase inhibitor | Phase II |
| [250] |
| Benzafibrate (+UDCA) | PPAR agonist | Phase III (BEZASCLER) |
| |
| Seladelpar | PPARδ agonist | Phase II (NCT04024813) |
| |
| Simvastatin | HMGCoA reductase inhibitors | Phase III (PiSCATIN) |
| |
| Vancomycin | Antibiotics | Phase III (NCT03710122) |
| |
| CM-101 | Monoclonal Ab blocking CCL24 | Phase II (SPRING) |
| |
| Cenicriviroc | Dual antagonist of CCR2 and CCR5 | Phase II (PERSEUS) |
| [251] |
| Timolumab | Monoclonal anti-VAP-1 antibody | Phase II (BUTEO) |
| |
| NGM282 | FGF19 analog | Phase II |
| [252] |
| Sulfasalazine | Aminosalicylates | Phase II (NCT02177136) |
| |
| Fecal Microbiota Transplantation | Restore the microbiome | Phase II |
| [253] |
| Umbilical Cord Mesenchymal Stem Cells | Repair of damaged tissue and immuno-modulation | Phase II (NCT03516006) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-W.; Kim, J.-H.; Kim, S.-E.; Jung, J.H.; Jang, M.-K.; Park, S.-H.; Lee, M.-S.; Kim, H.-S.; Suk, K.T.; Kim, D.J. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics. Biomedicines 2022, 10, 1288. https://doi.org/10.3390/biomedicines10061288
Park J-W, Kim J-H, Kim S-E, Jung JH, Jang M-K, Park S-H, Lee M-S, Kim H-S, Suk KT, Kim DJ. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics. Biomedicines. 2022; 10(6):1288. https://doi.org/10.3390/biomedicines10061288
Chicago/Turabian StylePark, Ji-Won, Jung-Hee Kim, Sung-Eun Kim, Jang Han Jung, Myoung-Kuk Jang, Sang-Hoon Park, Myung-Seok Lee, Hyoung-Su Kim, Ki Tae Suk, and Dong Joon Kim. 2022. "Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics" Biomedicines 10, no. 6: 1288. https://doi.org/10.3390/biomedicines10061288
APA StylePark, J.-W., Kim, J.-H., Kim, S.-E., Jung, J. H., Jang, M.-K., Park, S.-H., Lee, M.-S., Kim, H.-S., Suk, K. T., & Kim, D. J. (2022). Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics. Biomedicines, 10(6), 1288. https://doi.org/10.3390/biomedicines10061288