
Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Conditions
2.2. Drug Transporters Activity
2.3. Viability Assay
2.4. Cell Death Analysis
2.5. Apoptosis, DNA Damage, and Cell Proliferation Analysis
2.6. Cell Cycle Analysis
2.7. Statistical Analysis
3. Results
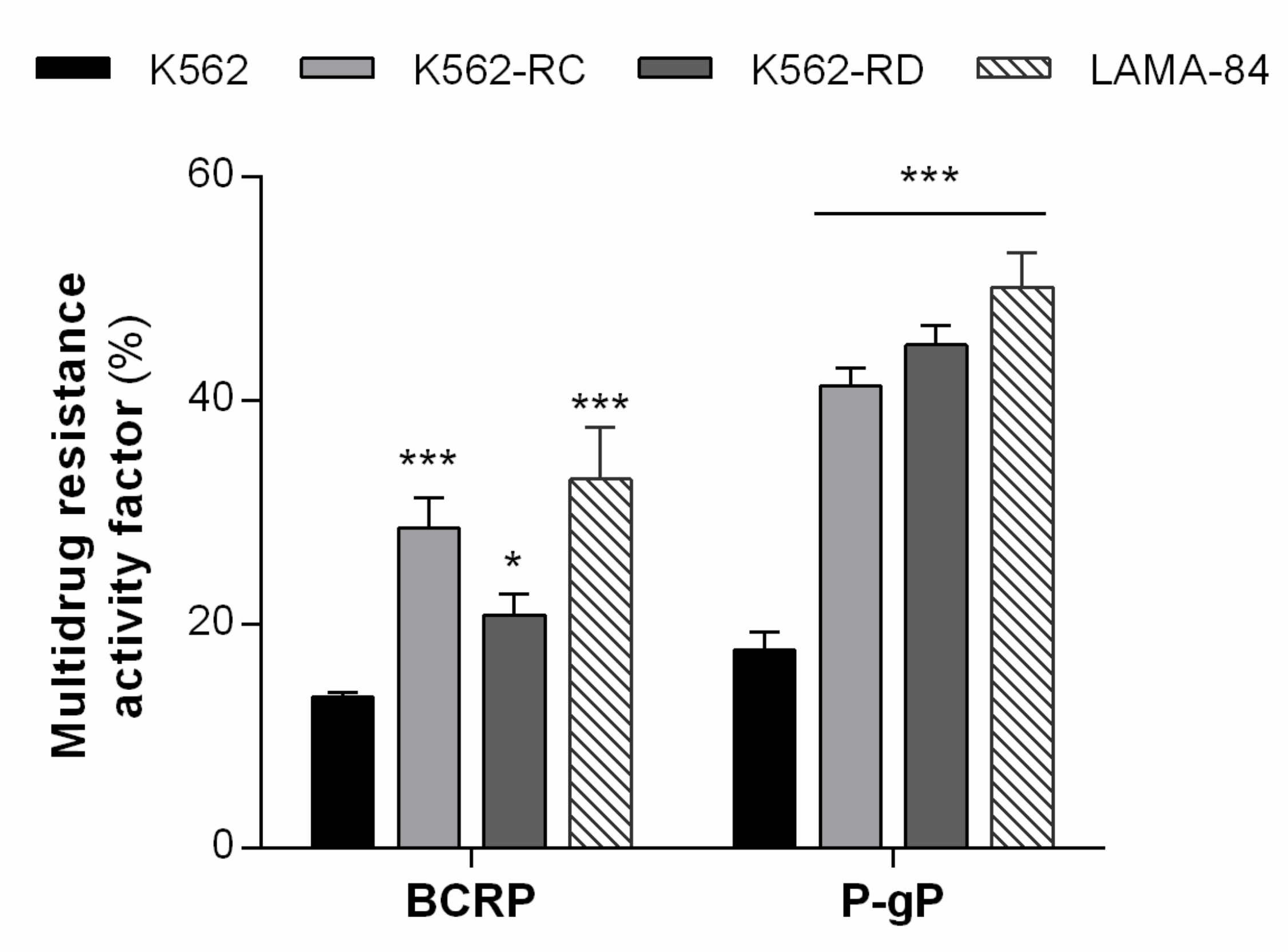
3.1. Resistant Cell Lines Showed MDR Phenotype Mediated by P-gP and BCRP Activity
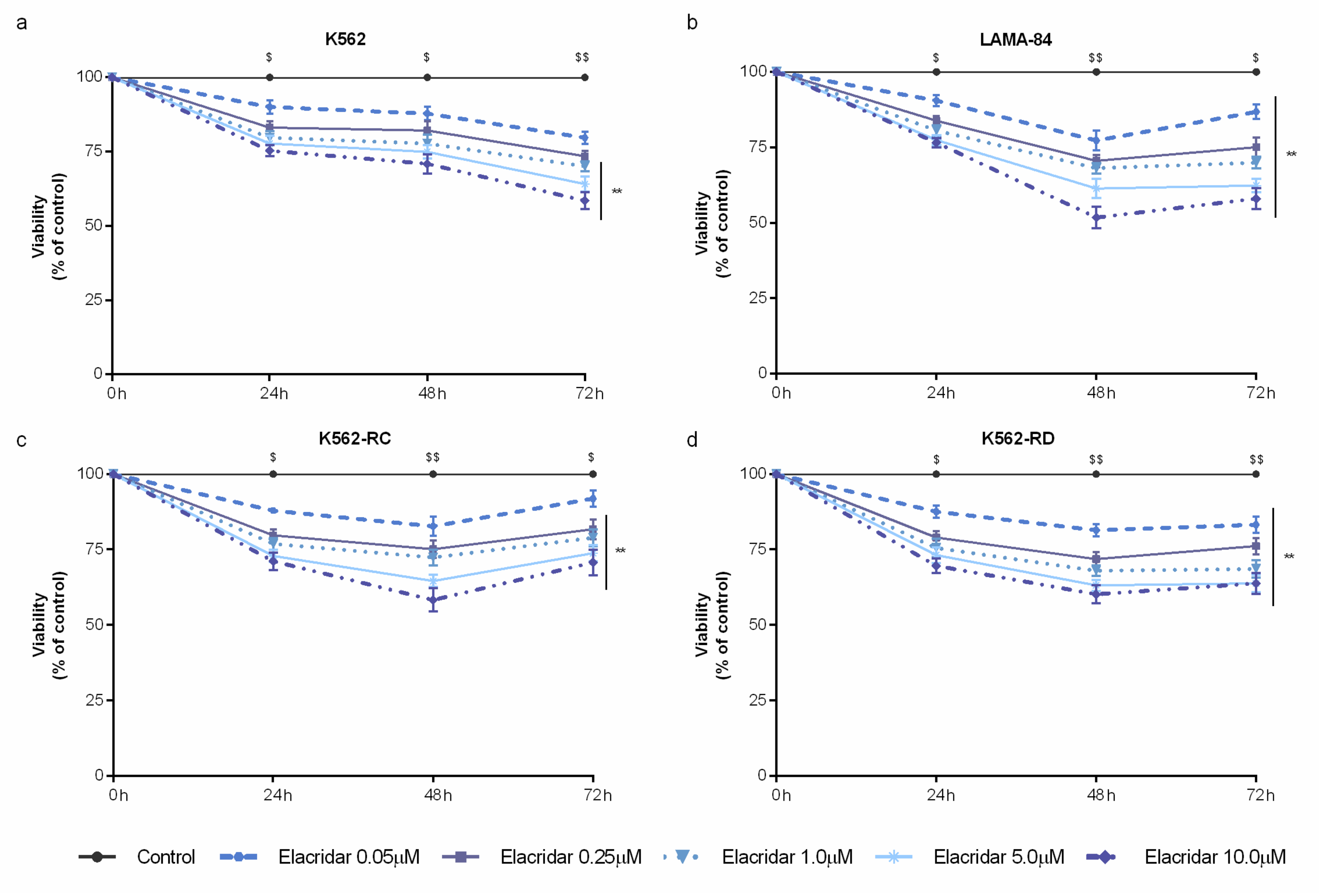
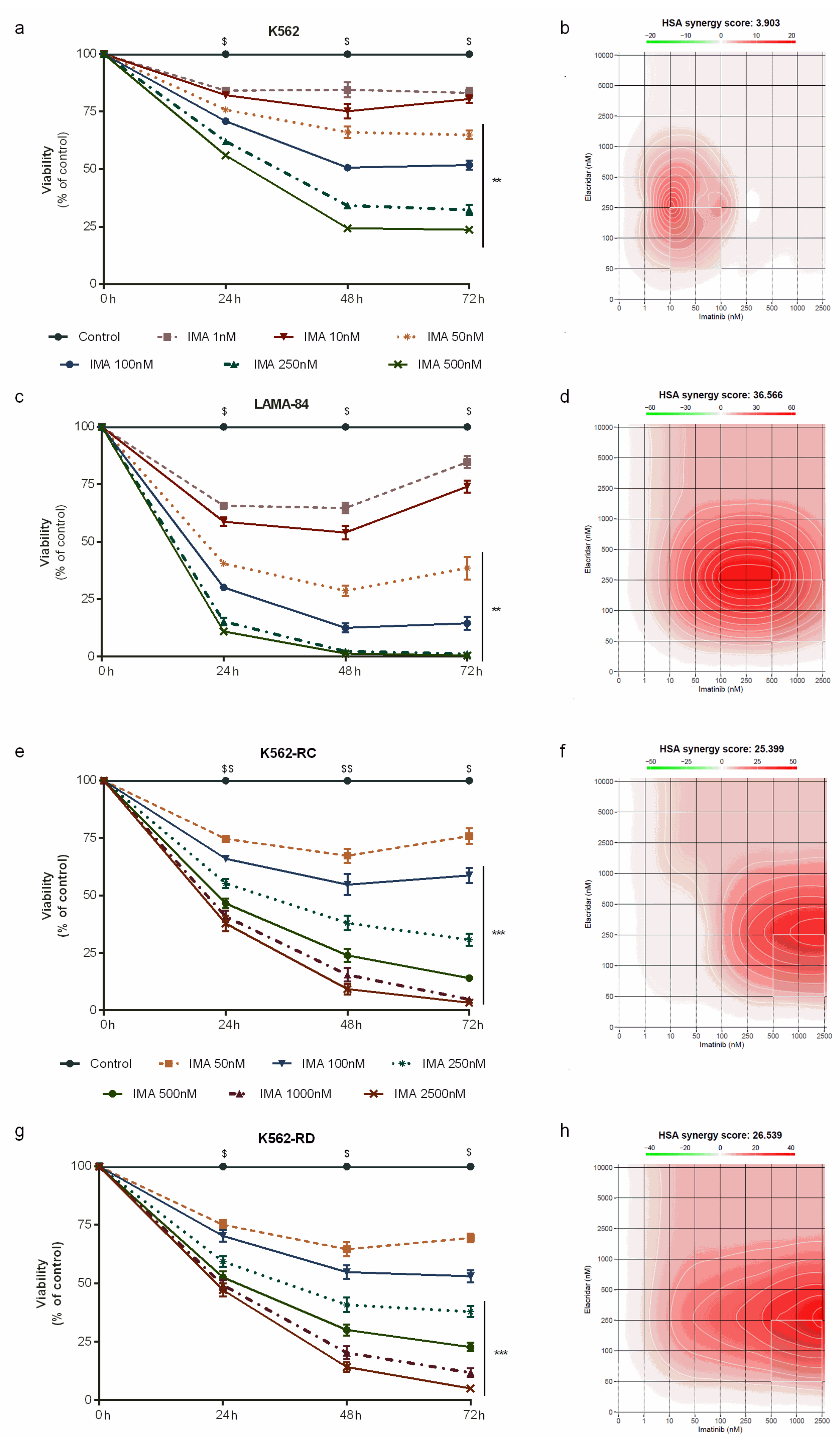
3.2. Combined Treatment with Elacridar Overcomes Imatinib Resistance
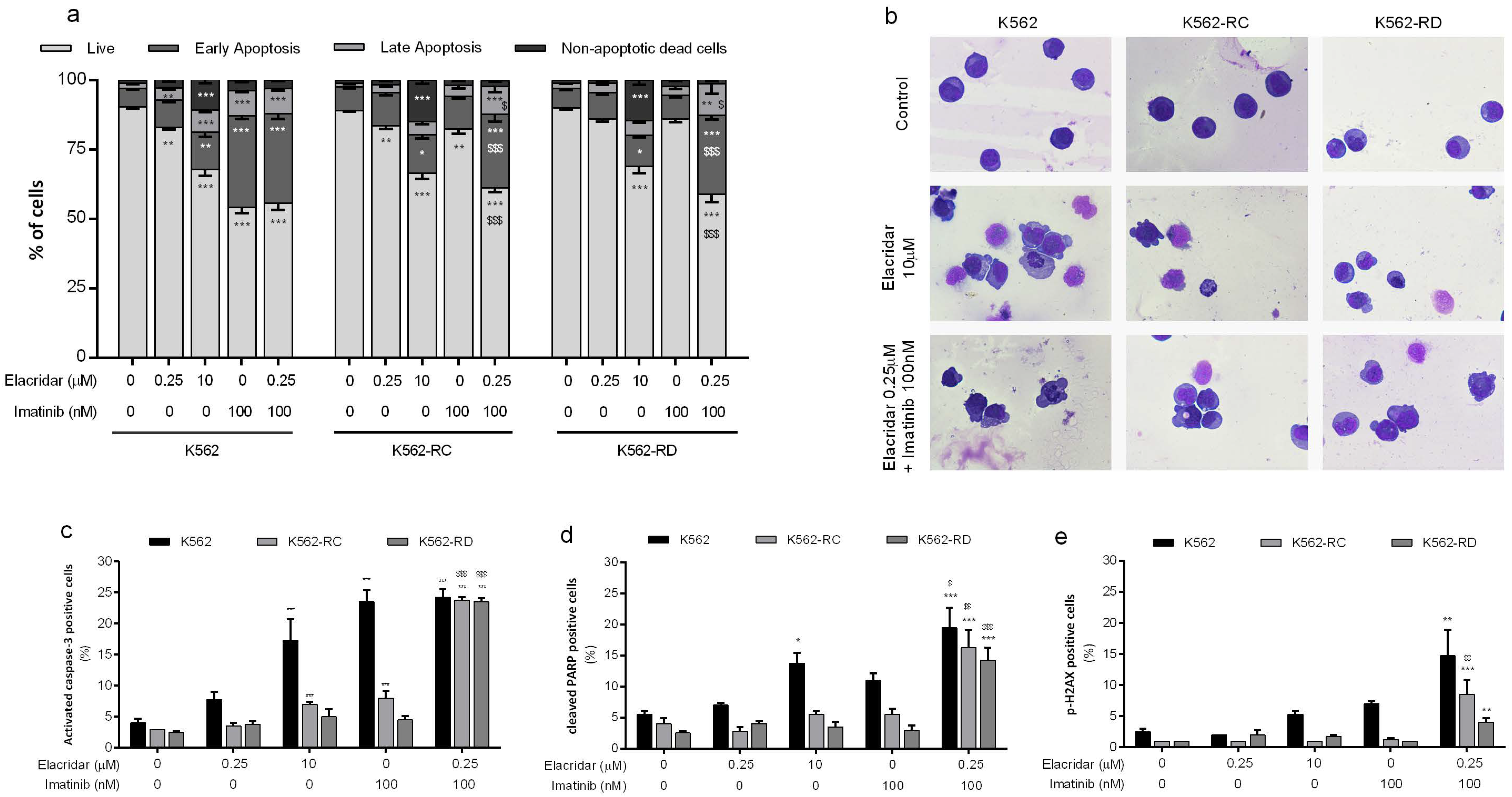
3.3. Apoptosis as the Main Mechanism of Cell Death in the Combined Treatment
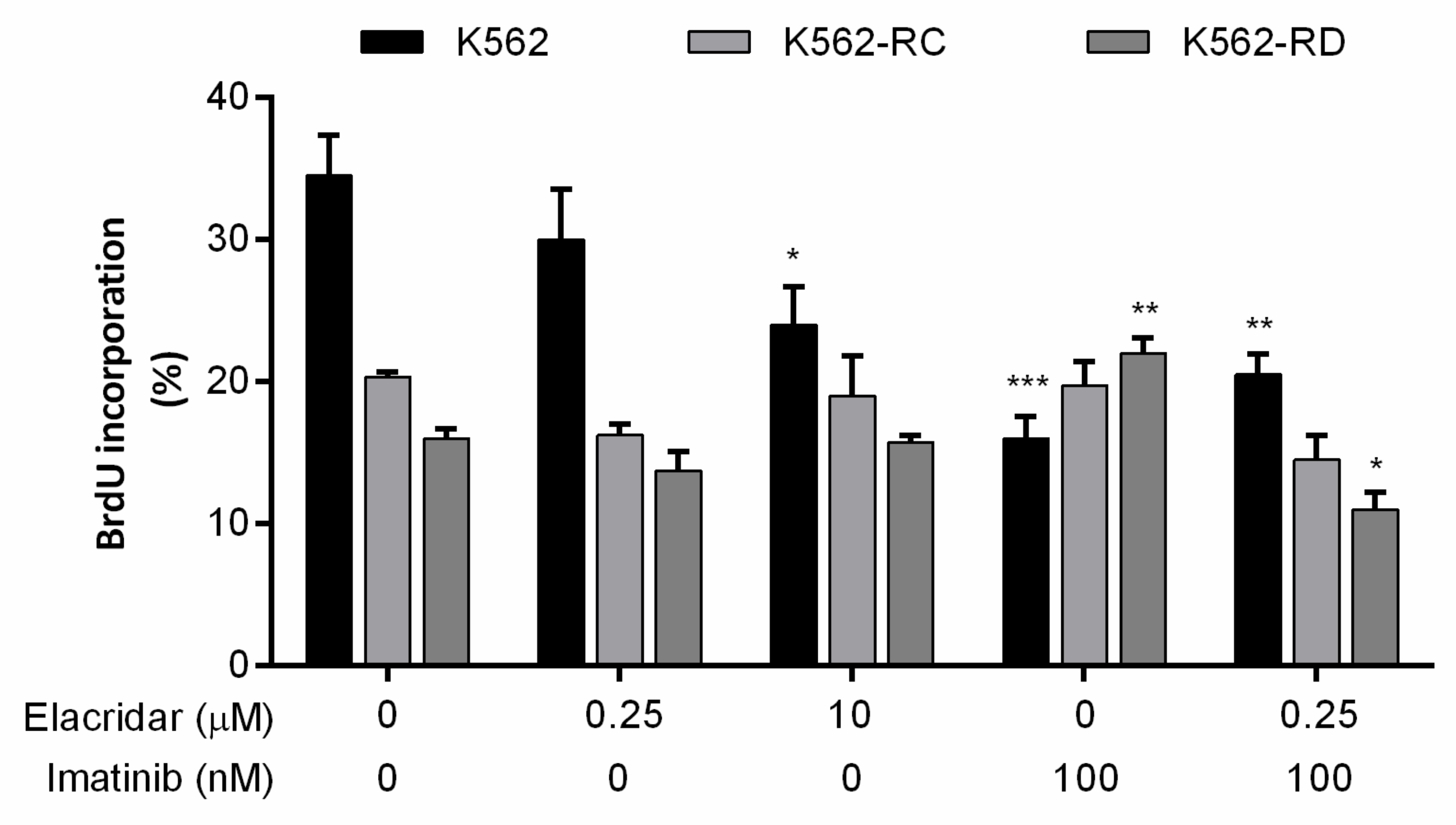
3.4. Drug Combination Reduces Cell Proliferation and Alters Cell Cycle Distribution
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 1–30. [Google Scholar] [CrossRef]
- Ali, M.A.M. Chronic Myeloid Leukemia in the Era of Tyrosine Kinase Inhibitors: An Evolving Paradigm of Molecularly Targeted Therapy. Mol. Diagn. Ther. 2016, 20, 315–333. [Google Scholar] [CrossRef]
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G. A review of the European LeukemiaNet recommendations for the management of CML. Ann. Hematol. 2015, 94, 141–147. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saussele, S.; Rosti, G.; Mahon, F.X.; Janssen, J.J.W.M.; Hjorth-Hansen, H.; Richter, J.; Buske, C. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2017, 28, 41–51. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia—From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S.V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2007, 22, 445–447. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, C.; Ozvegy-Laczka, C.; Apáti, A.; Magócsi, M.; Német, K.; Orfi, L.; Kéri, G.; Katona, M.; Takáts, Z.; Váradi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Hasanabady, M.H.; Kalalinia, F. ABCG2 inhibition as a therapeutic approach for overcoming multidrug resistance in cancer. J. Biosci. 2016, 41, 313–324. [Google Scholar] [CrossRef]
- da Cunha Vasconcelos, F.; Mauricio Scheiner, M.A.; Moellman-Coelho, A.; Mencalha, A.L.; Renault, I.Z.; Rumjanek, V.M.; Maia, R.C. Low ABCB1 and high OCT1 levels play a favorable role in the molecular response to imatinib in CML patients in the community clinical practice. Leuk. Res. 2016, 51, 3–10. [Google Scholar] [CrossRef]
- DeGorter, M.; Xia, C.; Yang, J.; Kim, R. Drug Transporters in Drug Efficacy and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, N.; Sharma, S. ATP binding cassette transporters and cancer: Revisiting their controversial role. Pharmacogenomics 2021, 22, 1211–1235. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remião, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Dong, J.; Qin, Z.; Zhang, W.-D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.-S.; Cheng, X.-D.; Qin, J.-J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updates 2020, 49, 100681. [Google Scholar] [CrossRef]
- Dash, R.P.; Jayachandra Babu, R.; Srinivas, N.R. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 915–933. [Google Scholar] [CrossRef]
- Sarisozen, C.; Vural, I.; Levchenko, T.; Hincal, A.A.; Torchilin, V.P. Long-circulating PEG-PE micelles co-loaded with paclitaxel and elacridar (GG918) overcome multidrug resistance. Drug Deliv. 2012, 19, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; He, J.; Li, X.; Li, B.; Zhang, L.; Wang, Y.; Cheng, D.; Shuai, X. Polymeric nanovesicles as simultaneous delivery platforms with doxorubicin conjugation and elacridar encapsulation for enhanced treatment of multidrug-resistant breast cancer. J. Mater. Chem. B 2018, 6, 7521–7529. [Google Scholar] [CrossRef]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.-M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Sato, H.; Siddig, S.; Uzu, M.; Suzuki, S.; Nomura, Y.; Kashiba, T.; Gushimiyagi, K.; Sekine, Y.; Uehara, T.; Arano, Y.; et al. Elacridar enhances the cytotoxic effects of sunitinib and prevents multidrug resistance in renal carcinoma cells. Eur. J. Pharmacol. 2015, 746, 258–266. [Google Scholar] [CrossRef]
- Chen, H.; Shien, K.; Suzawa, K.; Tsukuda, K.; Tomida, S.; Sato, H.; Torigoe, H.; Watanabe, M.; Namba, K.; Yamamoto, H.; et al. Elacridar, a third-generation ABCB1 inhibitor, overcomes resistance to docetaxel in non-small cell lung cancer. Oncol. Lett. 2017, 14, 4349–4354. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Kuppens, I.E.L.M.; Witteveen, E.O.; Jewell, R.C.; Radema, S.A.; Paul, E.M.; Mangum, S.G.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H.M. A Phase I, Randomized, Open-Label, Parallel-Cohort, Dose-Finding Study of Elacridar (GF120918) and Oral Topotecan in Cancer Patients. Clin. Cancer Res. 2007, 13, 3276–3285. [Google Scholar] [CrossRef] [Green Version]
- Planting, A.S.T.; Sonneveld, P.; van der Gaast, A.; Sparreboom, A.; van der Burg, M.E.L.; Luyten, G.P.M.; de Leeuw, K.; de Boer-Dennert, M.; Wissel, P.S.; Jewell, R.C.; et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2005, 55, 91–99. [Google Scholar] [CrossRef]
- Alves, R.; Fonseca, A.R.; Gonçalves, A.C.; Ferreira-Teixeira, M.; Lima, J.; Abrantes, A.M.; Alves, V.; Rodrigues-Santos, P.; Jorge, L.; Matoso, E.; et al. Drug transporters play a key role in the complex process of Imatinib resistance in vitro. Leuk. Res. 2015, 39, 355–360. [Google Scholar] [CrossRef]
- Seigneurin, D.; Champelovier, P.; Mouchiroud, G.; Berthier, R.; Leroux, D.; Prenant, M.; McGregor, J.; Starck, J.; Morle, F.; Micouin, C.; et al. Human chronic myeloid leukemic cell line with positive Philadelphia chromosome exhibits megakaryocytic and erythroid characteristics. Exp. Hematol. 1987, 15, 822–832. [Google Scholar] [PubMed]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Alves, J.; Alves da Silva, A.; Freitas-Tavares, P.; Nascimento Costa, J.M.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Everolimus in combination with Imatinib overcomes resistance in Chronic myeloid leukaemia. Med. Oncol. 2019, 36, 1–10. [Google Scholar] [CrossRef]
- Jorge, J.; Petronilho, S.; Alves, R.; Coucelo, M.; Gonçalves, A.C.; Nascimento Costa, J.M.; Sarmento-Ribeiro, A.B. Apoptosis induction and cell cycle arrest of pladienolide B in erythroleukemia cell lines. Investig. New Drugs 2020, 38, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Eadie, L.N.; Dang, P.; Saunders, V.A.; Yeung, D.T.; Osborn, M.P.; Grigg, A.P.; Hughes, T.P.; White, D.L. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia 2016, 31, 75–82. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Huang, F.-F.; Wu, D.-S.; Zhang, L.; Yu, Y.-H.; Yuan, X.-Y.; Li, W.-J.; Chen, X.-P.; Zhao, X.-L.; Chen, F.-P.; Zeng, H. Inactivation of PTEN increases ABCG2 expression and the side population through the PI3K/Akt pathway in adult acute leukemia. Cancer Lett. 2013, 336, 96–105. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, F.E. CML stem cells: Evasion for better invasion. Blood 2017, 129, 141–142. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; O'Hare, T.; Deininger, M.W. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol. Oncol. Clin. N. Am. 2017, 31, 589–612. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Ward, K.W.; Azzarano, L.M. Preclinical Pharmacokinetic Properties of the P-Glycoprotein Inhibitor GF120918A (HCl salt of GF120918, 9,10-Dihydro-5-methoxy-9-oxo-[4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]phenyl]-4-acridine-carboxamide) in the Mouse, Rat, Dog, and Monkey. J. Pharmacol. Exp. Ther. 2004, 310, 703–709. [Google Scholar] [CrossRef]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. Oral coadministration of elacridar and ritonavir enhances brain accumulation and oral availability of the novel ALK/ROS1 inhibitor lorlatinib. Eur. J. Pharm. Biopharm. 2019, 136, 120–130. [Google Scholar] [CrossRef]
- Karbownik, A.; Sobańska, K.; Płotek, W.; Grabowski, T.; Klupczynska, A.; Plewa, S.; Grześkowiak, E.; Szałek, E. The influence of the coadministration of the p-glycoprotein modulator elacridar on the pharmacokinetics of lapatinib and its distribution in the brain and cerebrospinal fluid. Investig. New Drugs 2020, 38, 574–583. [Google Scholar] [CrossRef] [Green Version]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.M.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef]
- De Bruin, M.; Miyake, K.; Litman, T.; Robey, R.; Bates, S.E. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999, 146, 117–126. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Kelmansky, D.; Assaraf, Y.G.; Livney, Y.D. β-Casein micelles for oral delivery of SN-38 and elacridar to overcome BCRP-mediated multidrug resistance in gastric cancer. Eur. J. Pharm. Biopharm. 2018, 133, 240–249. [Google Scholar] [CrossRef]
- Stordal, B.; Hamon, M.; McEneaney, V.; Roche, S.; Gillet, J.-P.; O'Leary, J.J.; Gottesman, M.; Clynes, M. Resistance to paclitaxel in a cisplatin-resistant ovarian cancer cell line is mediated by P-glycoprotein. PLoS ONE 2012, 7, 0040717. [Google Scholar] [CrossRef] [Green Version]
- Hyafil, F.; Vergely, C.; Du Vignaud, P.; Grand-Perret, T. In Vitro and in Vivo Reversal of Multidrug Resistance by GF120918, an Acridonecarboxamide Derivative. Cancer Res. 1993, 53, 4595–4602. [Google Scholar]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G0/G1 (%) | S (%) | G2/M (%) | ||
|---|---|---|---|---|
| K562 Cells | ||||
| Control | 39.7 ± 0.7 | 49.0 ± 0.6 | 11.8 ± 0.9 | |
| Elacridar 0.25 μM | 37.0 ± 1.4 | 51.3 ± 0.7 | 11.7 ± 1.7 | |
| Elacridar 10 μM | 41.2 ± 1.6 | 41.8 ± 1.6 ** | 17.0 ± 1.8 | |
| IMA 100 ηM | 38.3 ± 2.1 | 47.3 ± 0.8 | 14.3 ± 1.7 | |
| Elacridar 0.25 μM + IMA 100 ηM | 46.2 ± 2.7 $ | 41.3 ± 2.4 **,$ | 12.5 ± 2.3 | |
| K562-RC Cells | ||||
| Control | 67.3 ± 2.2 | 22.5 ± 1.4 | 10.2 ± 0.8 | |
| Elacridar 0.25 μM | 65.2 ± 3.2 | 23.8 ± 2.2 | 11.0 ± 1.0 | |
| Elacridar 10 μM | 62.5 ± 3.4 | 23.5 ± 1.6 | 14.0 ± 1.9 | |
| IMA 100 ηM | 51.8 ± 2.2 ** | 38.2 ± 0.9 *** | 10.0 ± 1.3 | |
| Elacridar 0.25 μM + IMA 100 ηM | 52.7 ± 1.9 ** | 37.5 ± 1.4 *** | 9.8 ± 0.8 | |
| K562-RD Cells | ||||
| Control | 63.2 ± 1.2 | 26.8 ± 1.1 | 10.2 ± 0.3 | |
| Elacridar 0.25 μM | 60.0 ± 2.3 | 29.0 ± 2.1 | 11.0 ± 0.4 | |
| Elacridar 10 μM | 63.8 ± 2.4 | 23.3 ± 2.1 | 13.0 ± 0.4 | |
| IMA 100 ηM | 51.3 ± 2.1 *** | 37.8 ± 1.9 *** | 10.8 ± 0.4 | |
| Elacridar 0.25 μM + IMA 100 ηM | 54.5 ± 1.7 * | 32.7 ± 1.2 | 12.8 ± 1.2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, R.; Gonçalves, A.C.; Jorge, J.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia. Biomedicines 2022, 10, 1158. https://doi.org/10.3390/biomedicines10051158
Alves R, Gonçalves AC, Jorge J, Almeida AM, Sarmento-Ribeiro AB. Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia. Biomedicines. 2022; 10(5):1158. https://doi.org/10.3390/biomedicines10051158
Chicago/Turabian StyleAlves, Raquel, Ana Cristina Gonçalves, Joana Jorge, António M. Almeida, and Ana Bela Sarmento-Ribeiro. 2022. "Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia" Biomedicines 10, no. 5: 1158. https://doi.org/10.3390/biomedicines10051158
APA StyleAlves, R., Gonçalves, A. C., Jorge, J., Almeida, A. M., & Sarmento-Ribeiro, A. B. (2022). Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia. Biomedicines, 10(5), 1158. https://doi.org/10.3390/biomedicines10051158