
Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Cell Counting and Cell Cycle Analysis
2.3. Antibodies
2.4. Western Blotting Analysis
2.5. Immunofluorescence Assay
2.6. 3D-Spheroid Forming Assay
2.7. DiD Proliferation Assay
2.8. Co-Immunoprecipitation Assay
2.9. Small Interference RNA Transfection
2.10. Bioinformatic Analysis
2.11. Statistical Analysis
3. Results
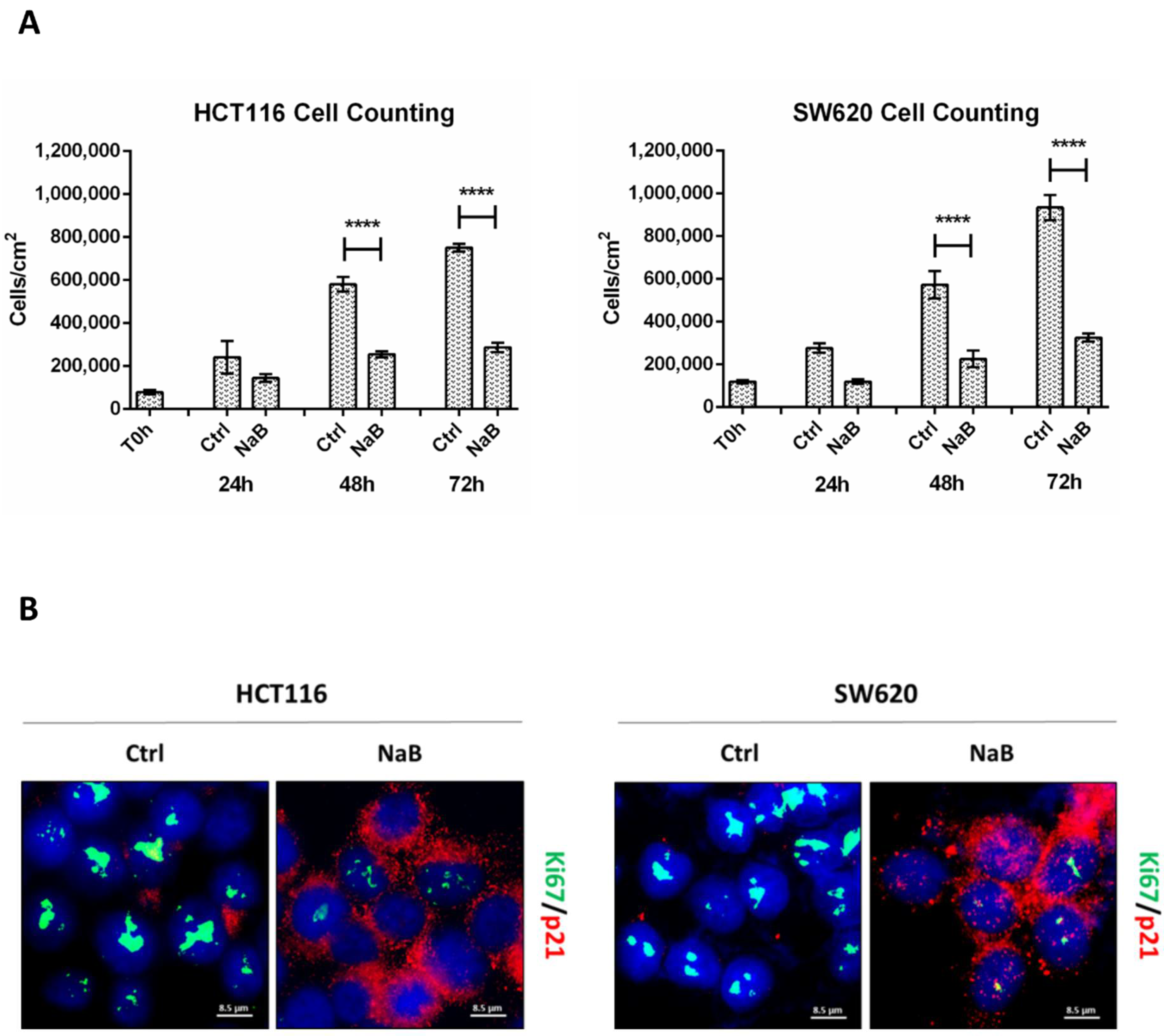
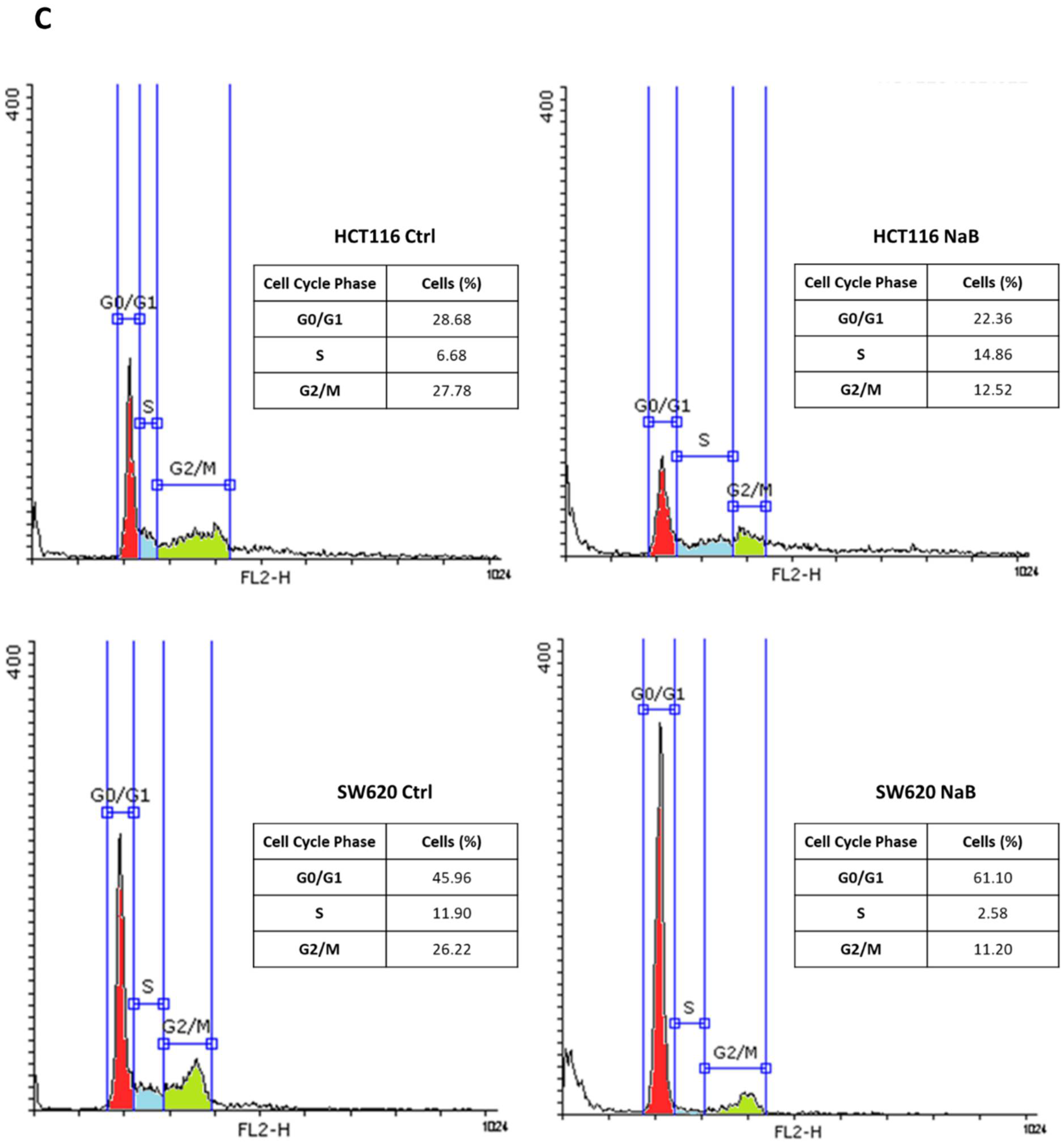
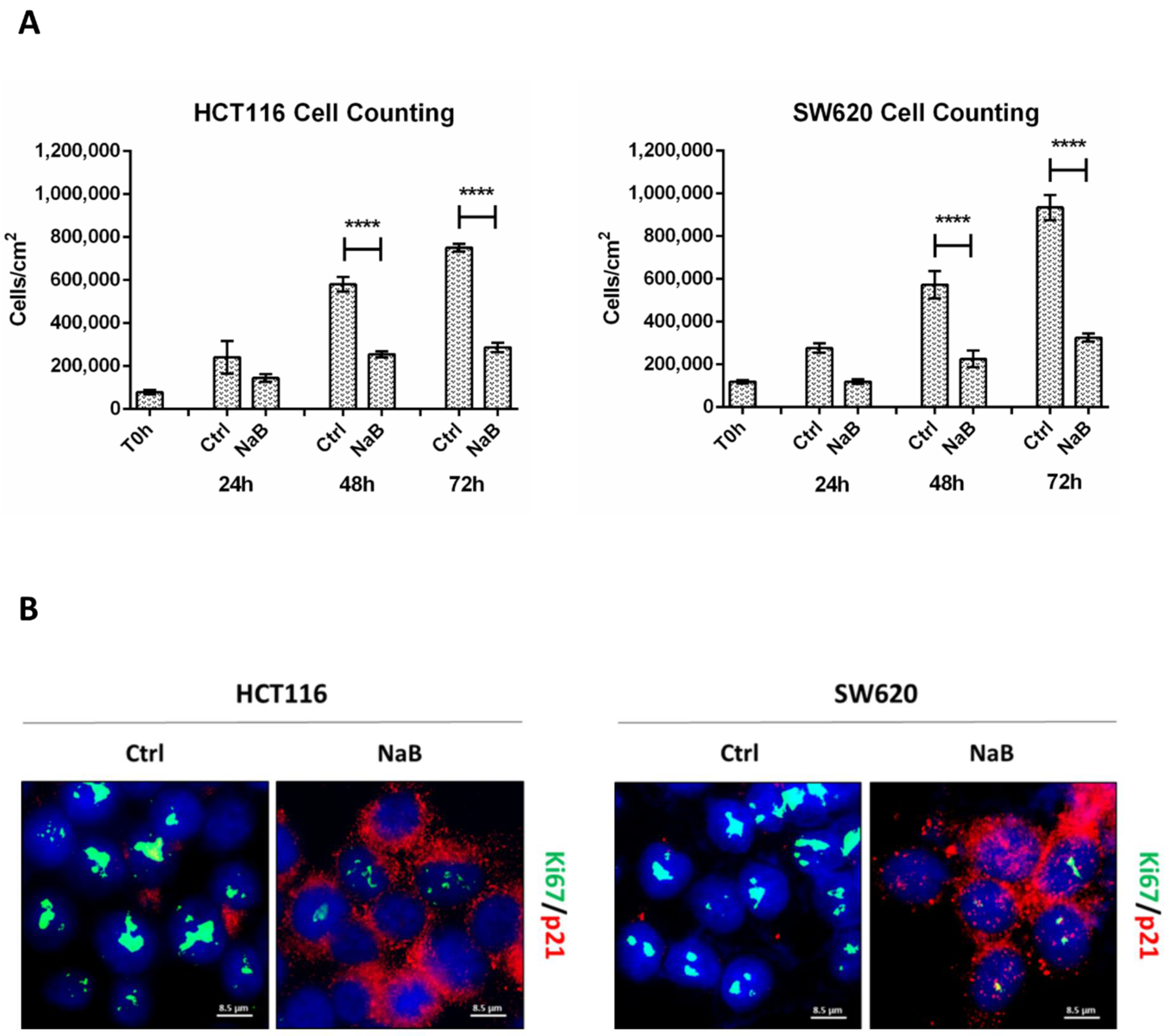
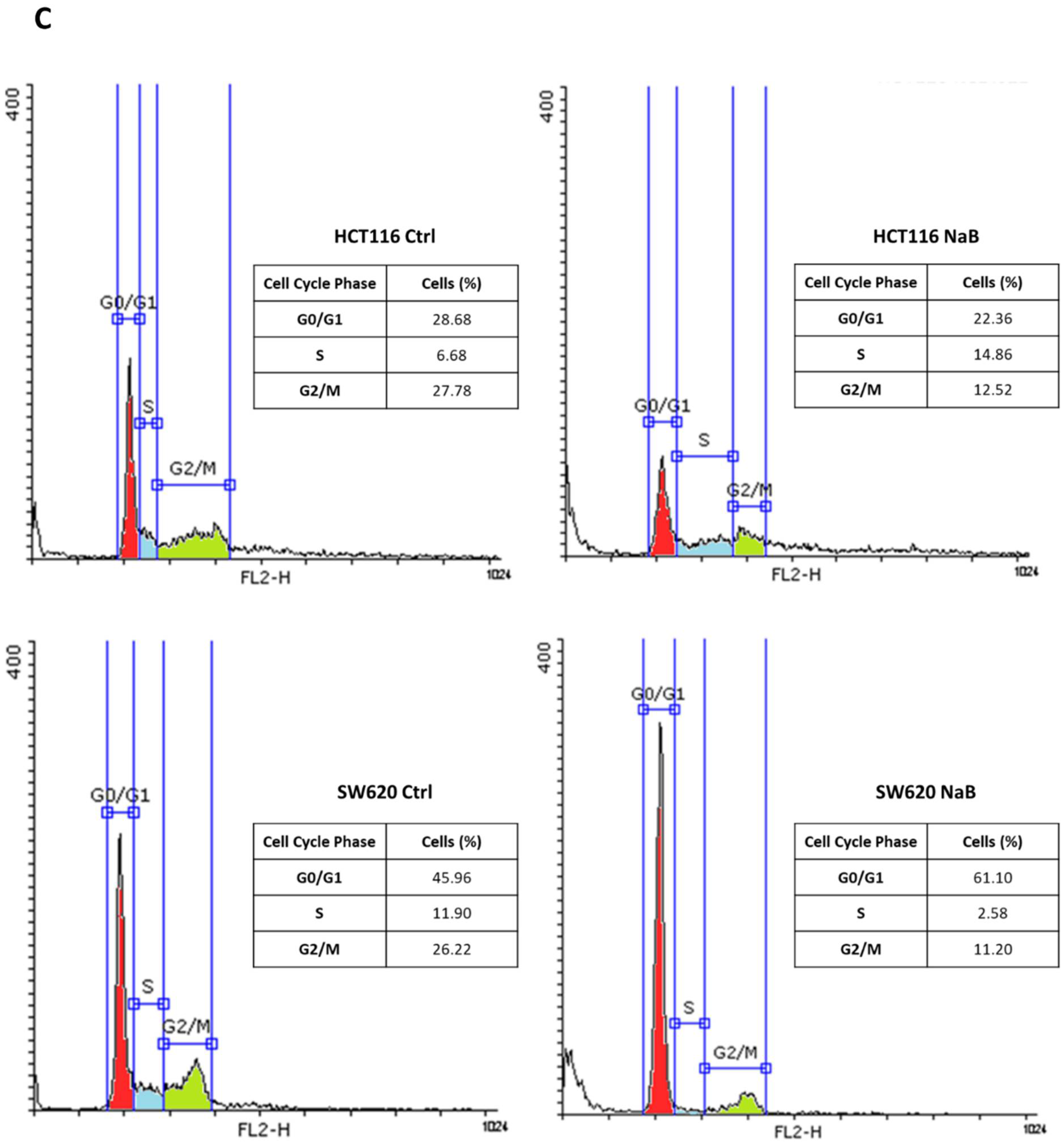
3.1. Butyrate Induces CRC Cell Growth Arrest
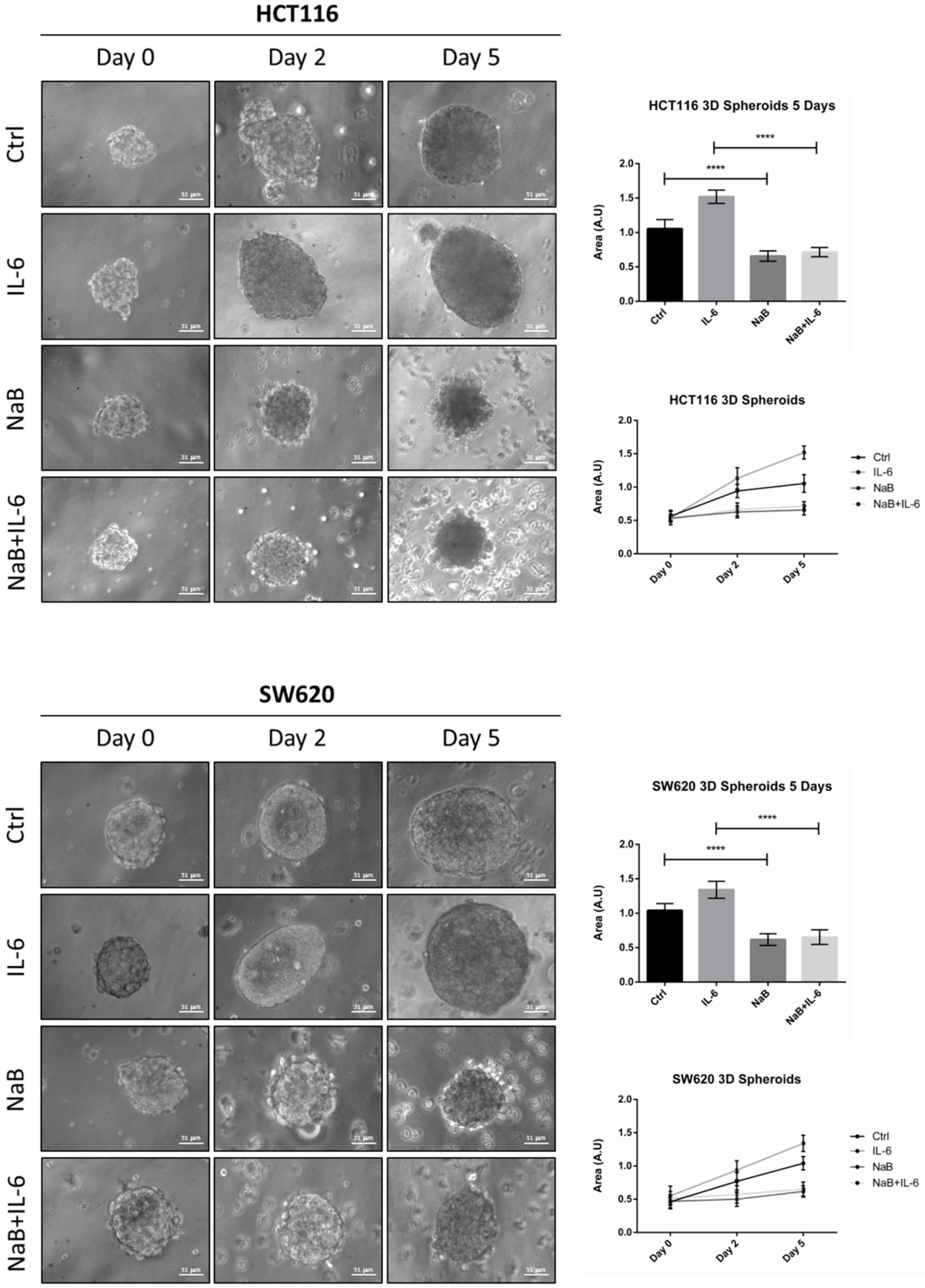
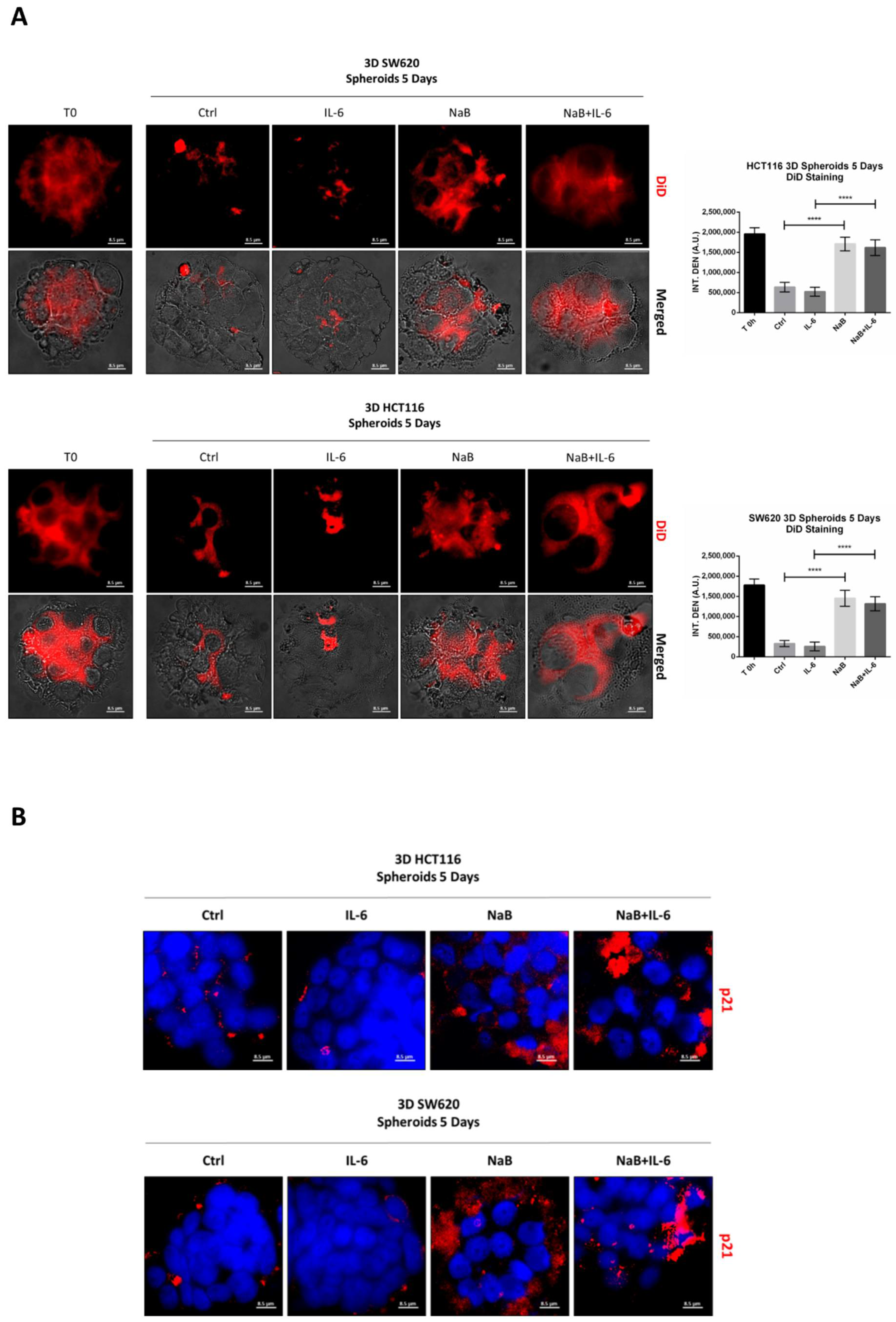
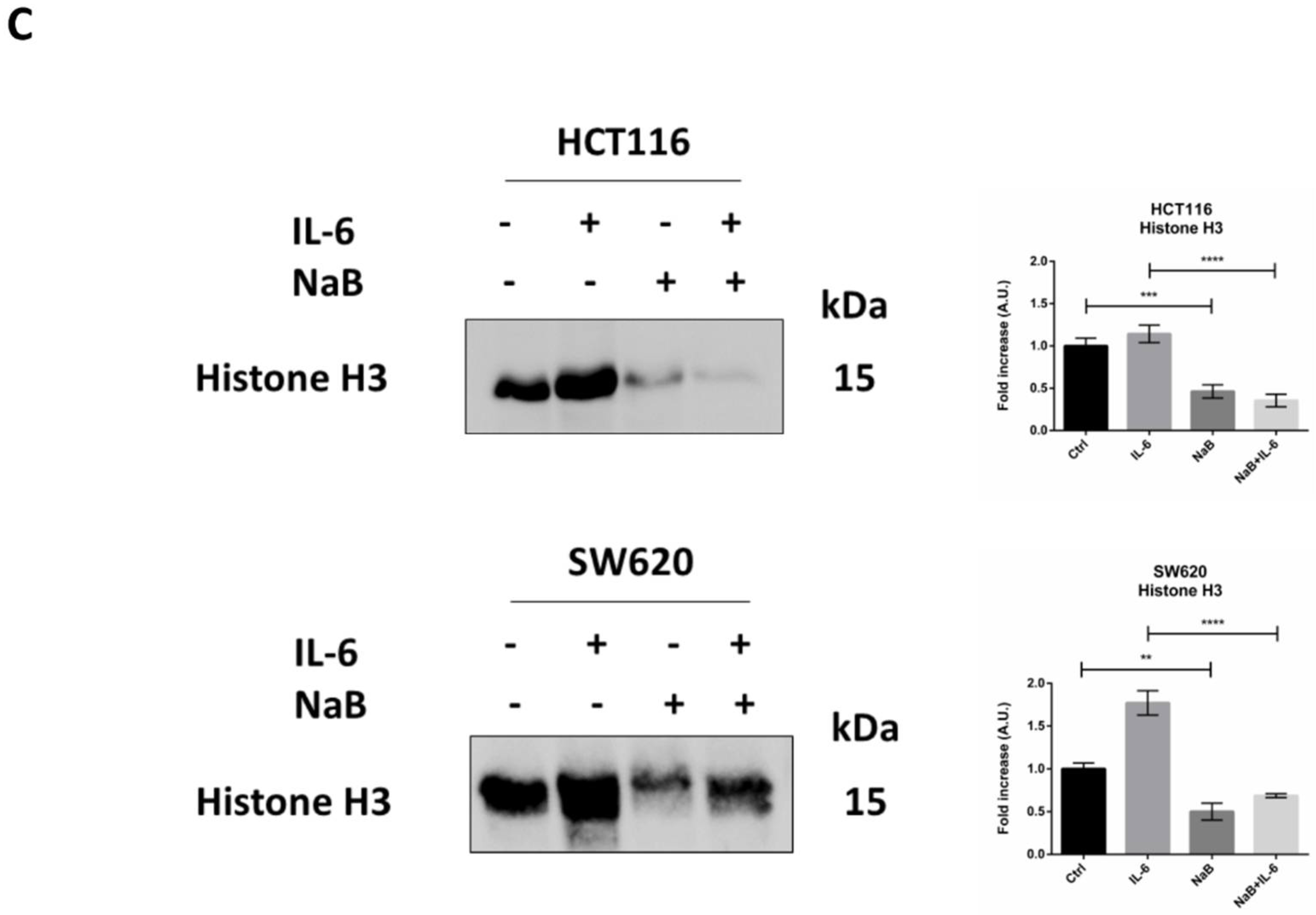
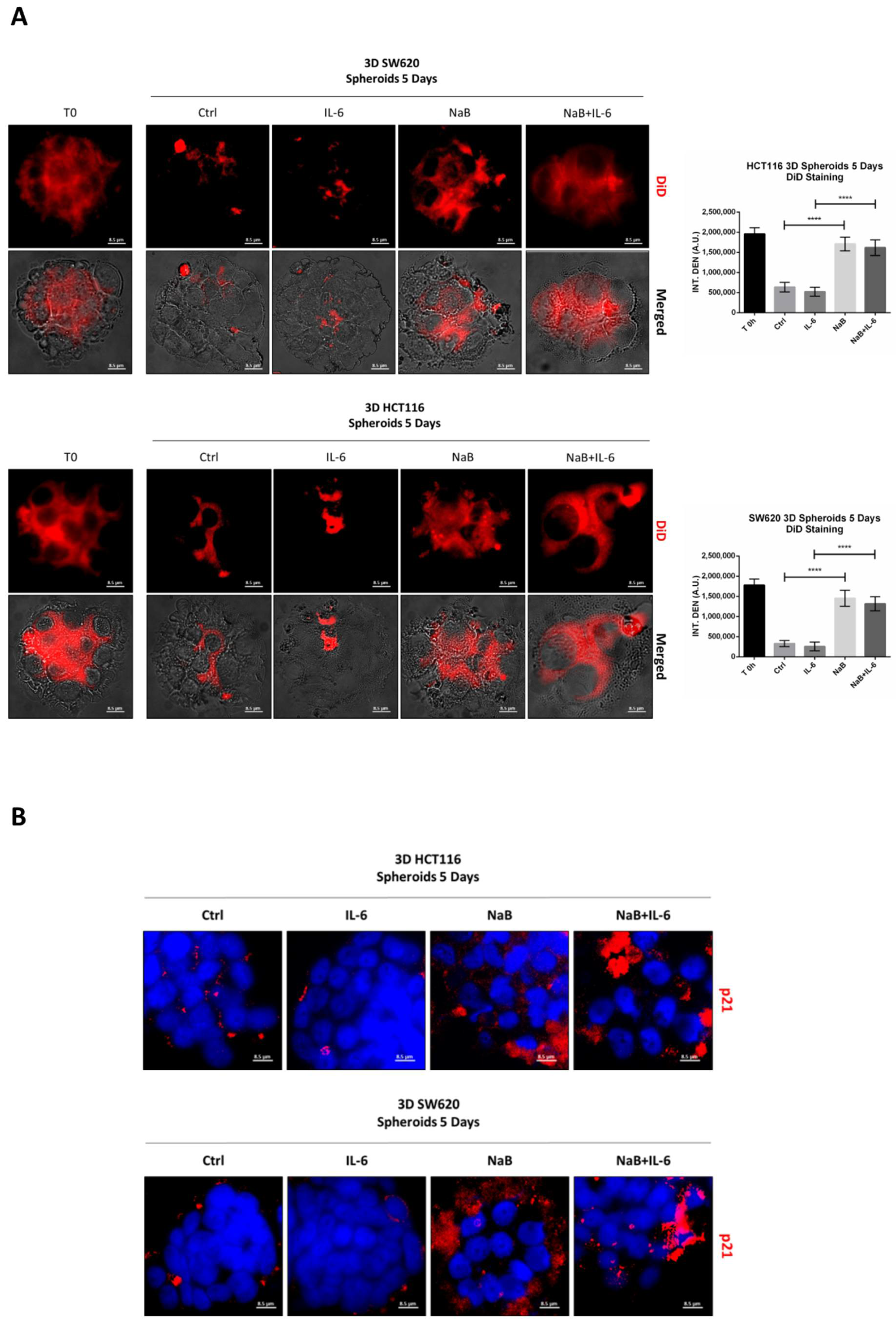
3.2. Butyrate Counteracts IL-6-Induced 3D Colorectal Cancer Spheroid Growth
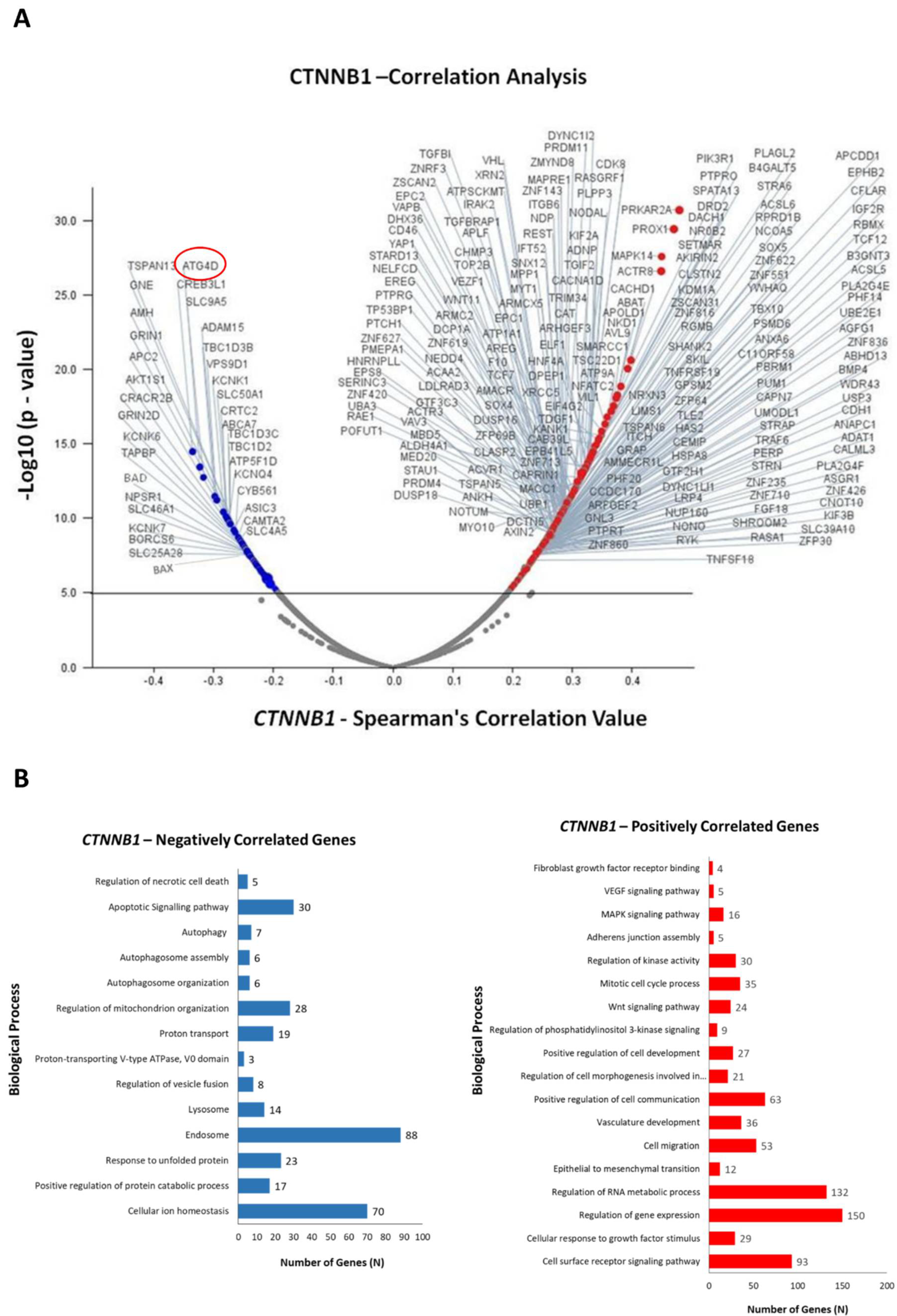
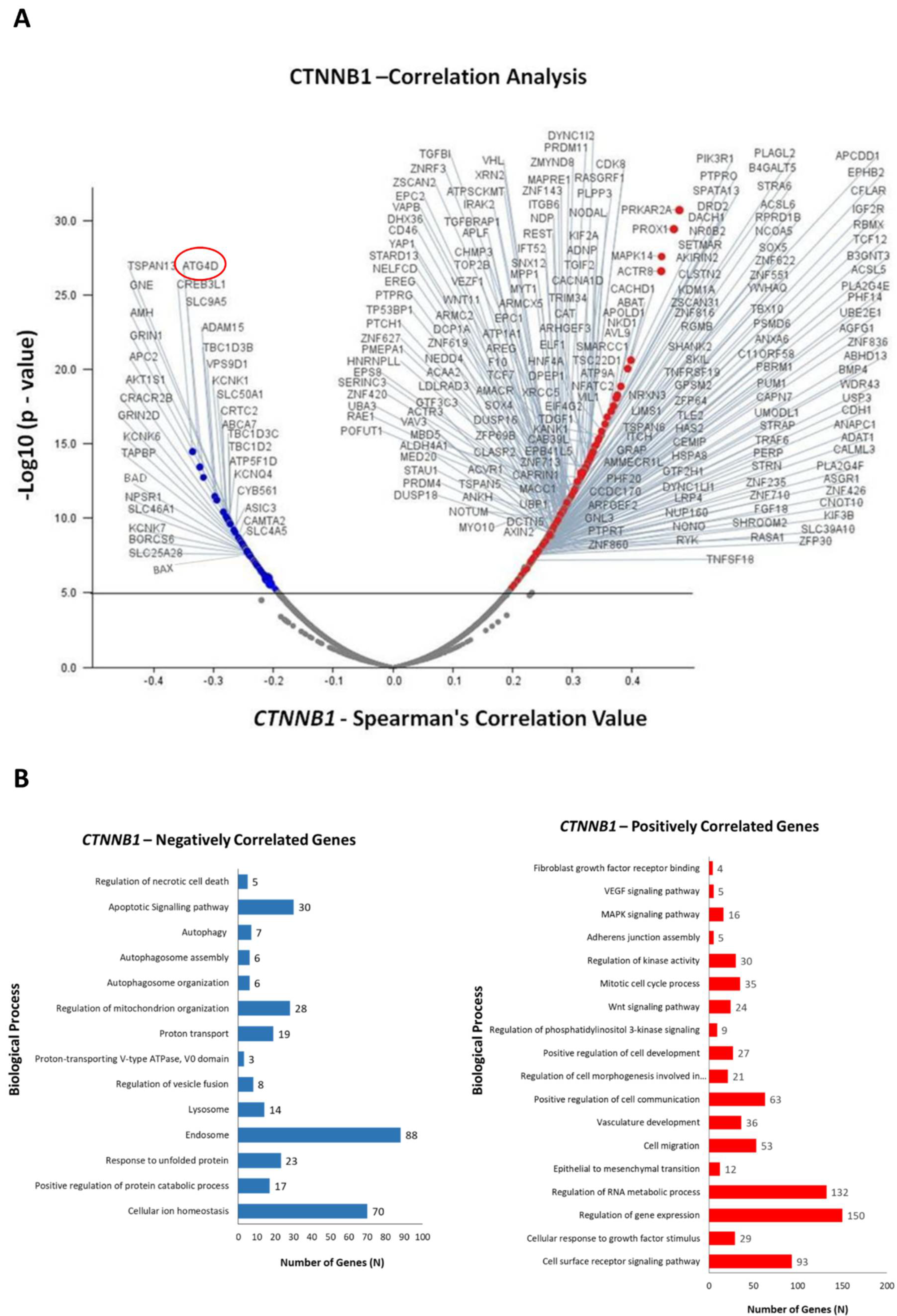
3.3. Identification of Differentially Expressed Genes in Correlation with CTNNB1
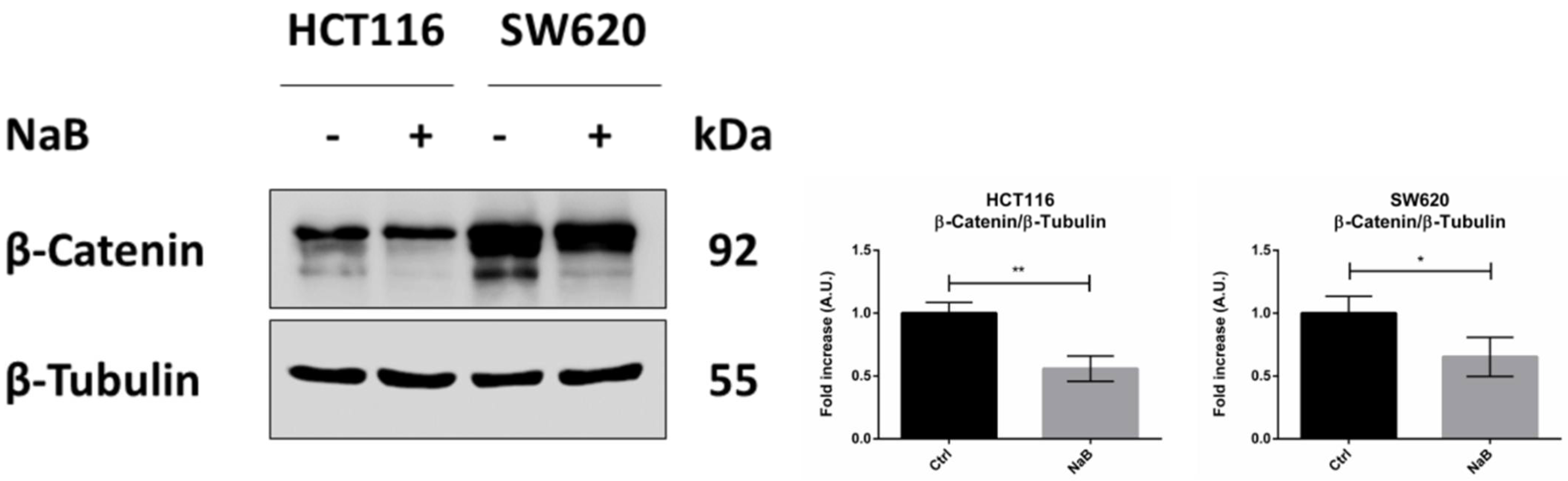
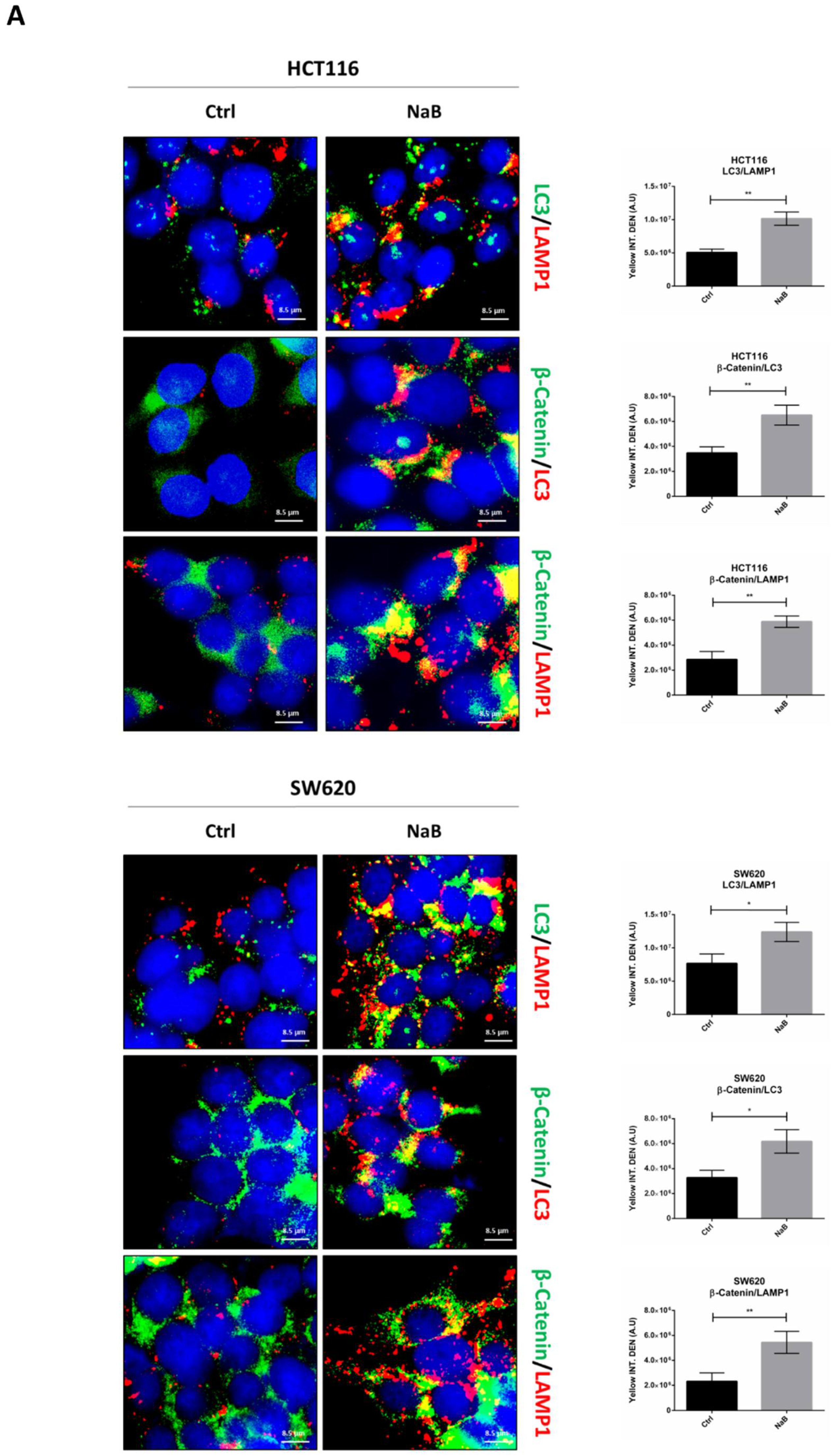
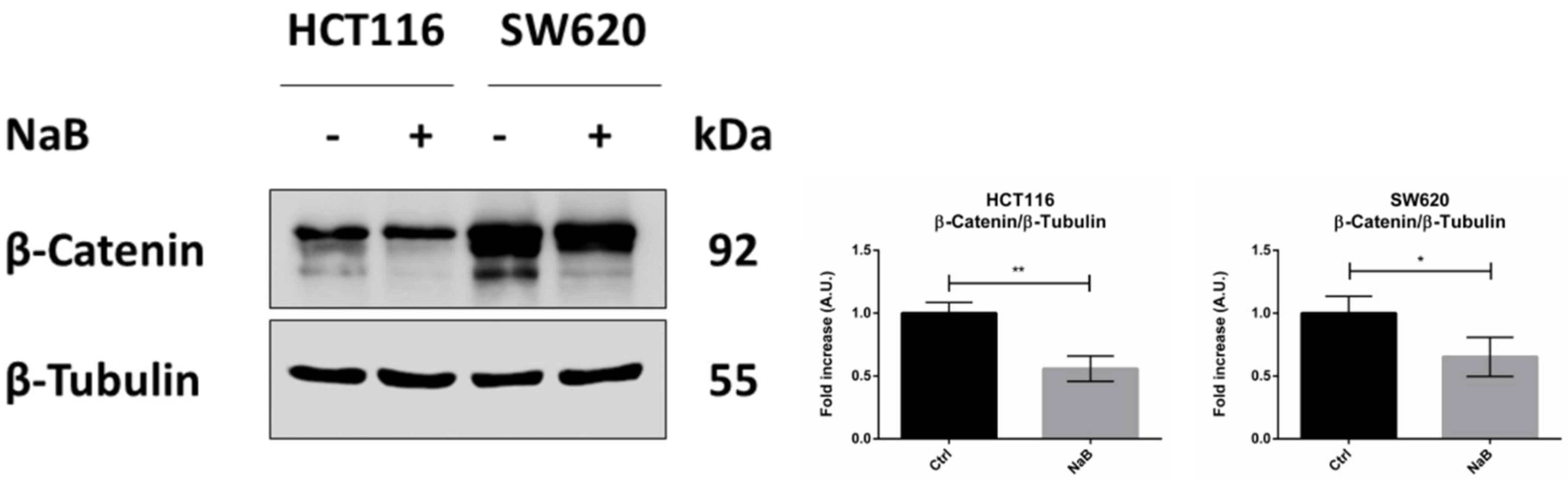
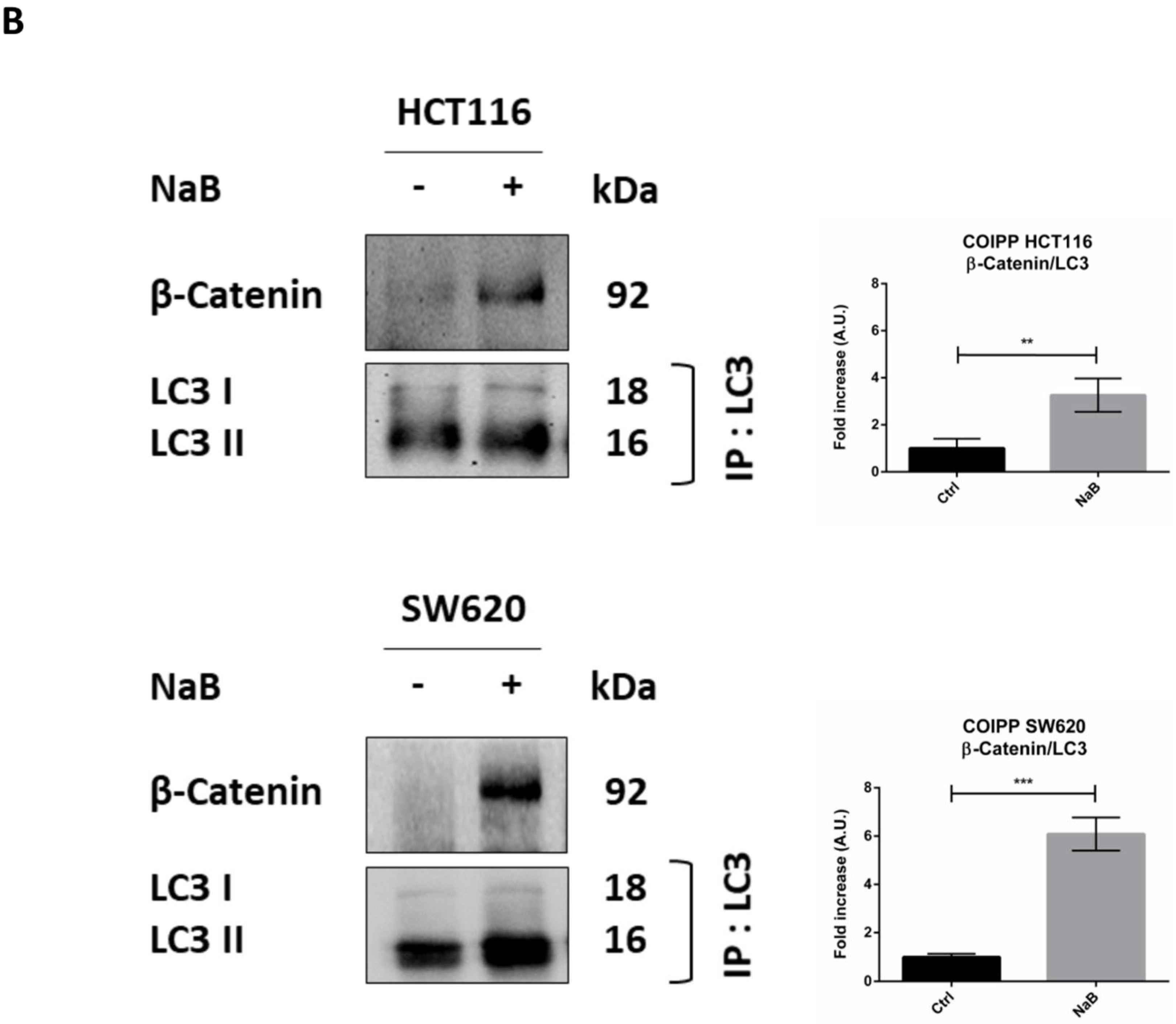
3.4. Butyrate Induces the Autophagosomal Sequestration and Subsequent Degradation of β-Catenin by Promoting Its Interaction with LC3
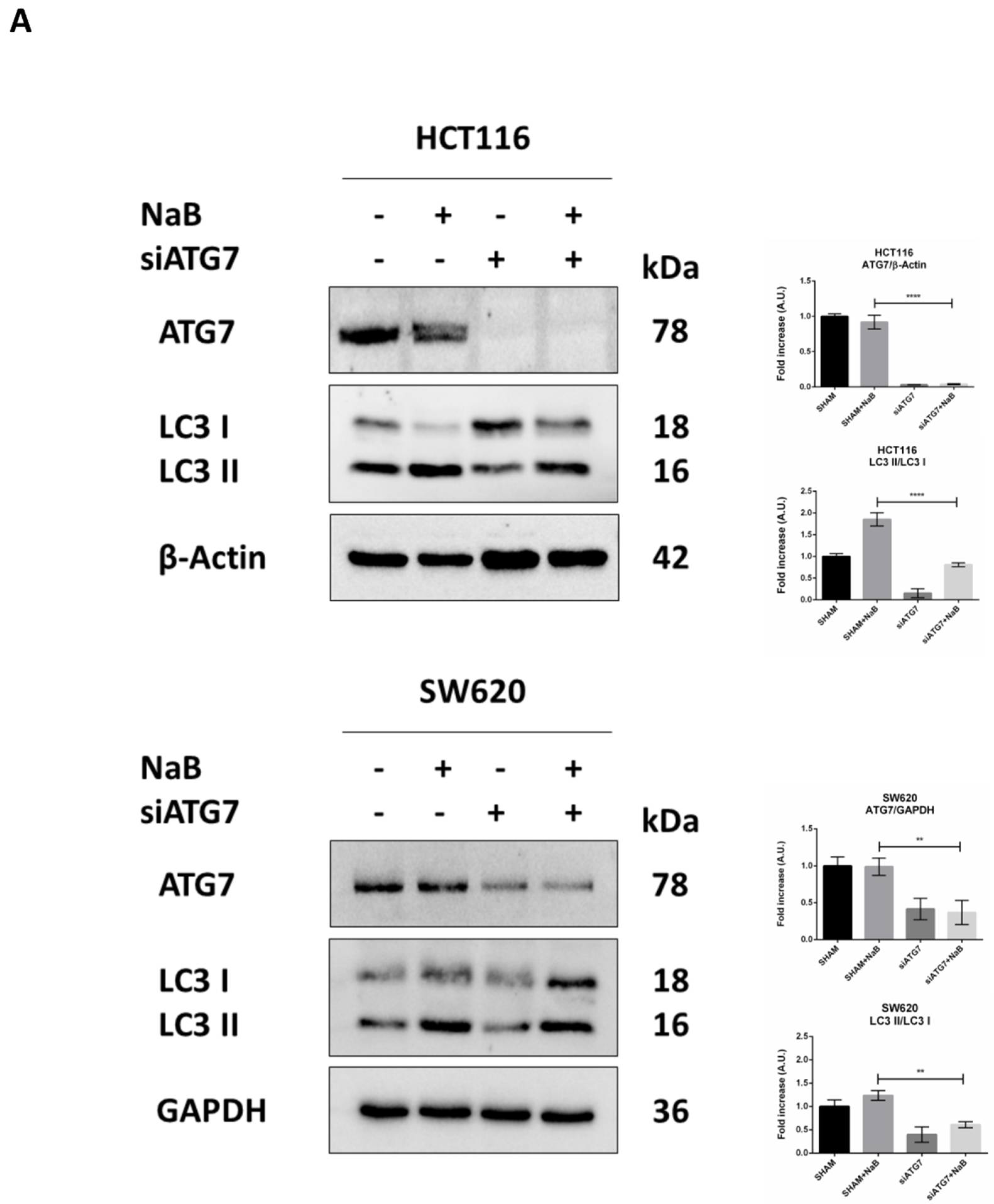
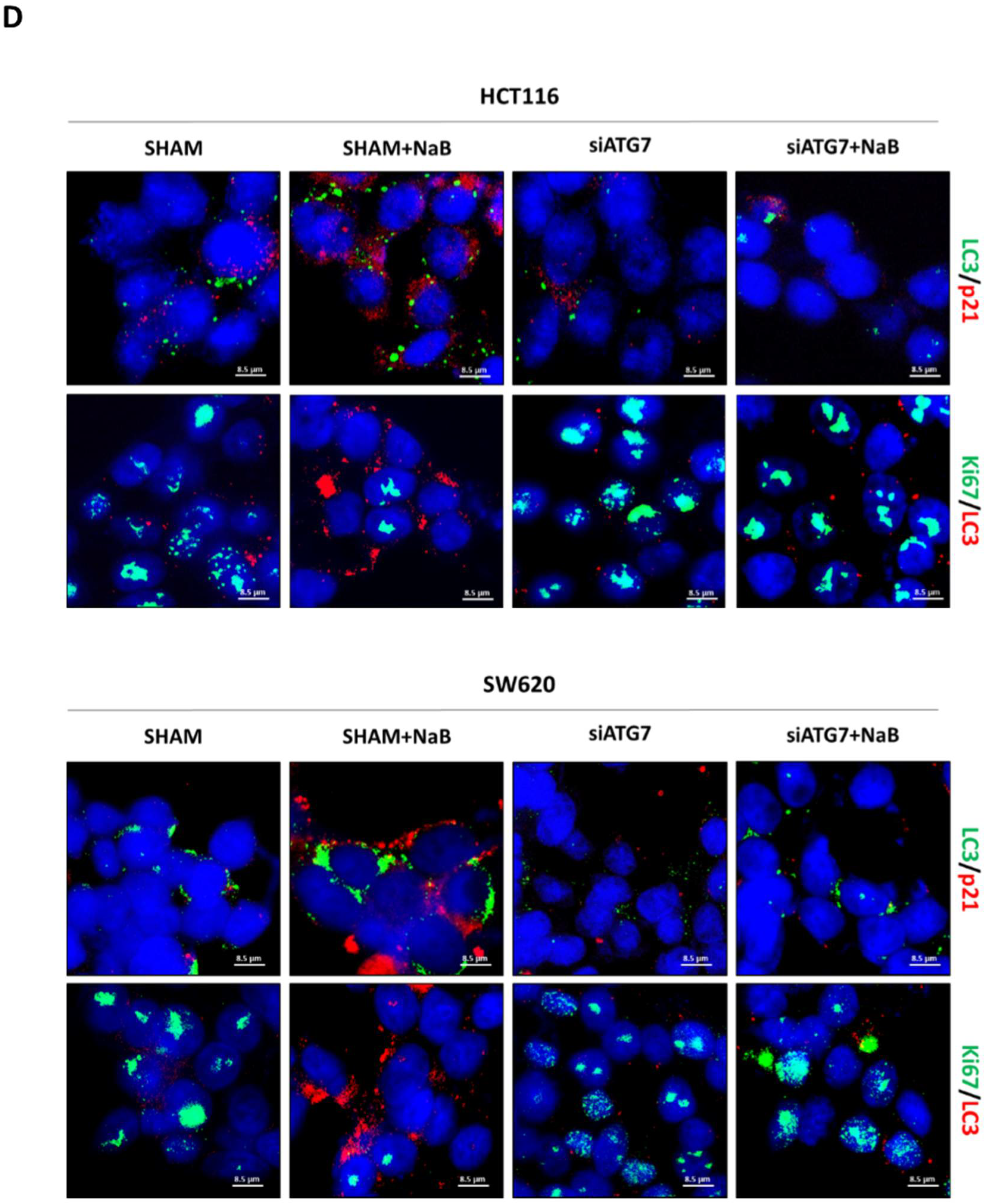
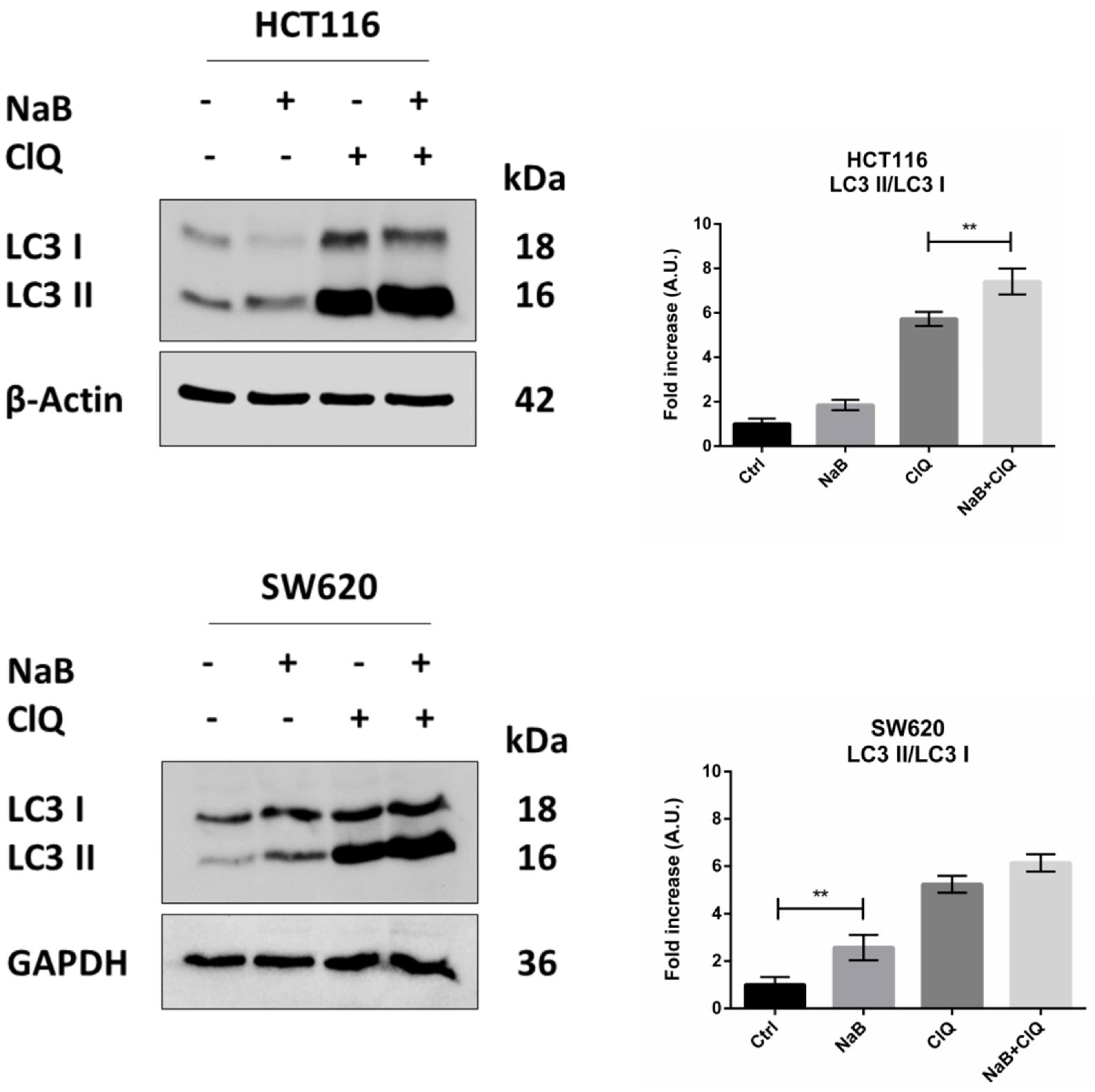
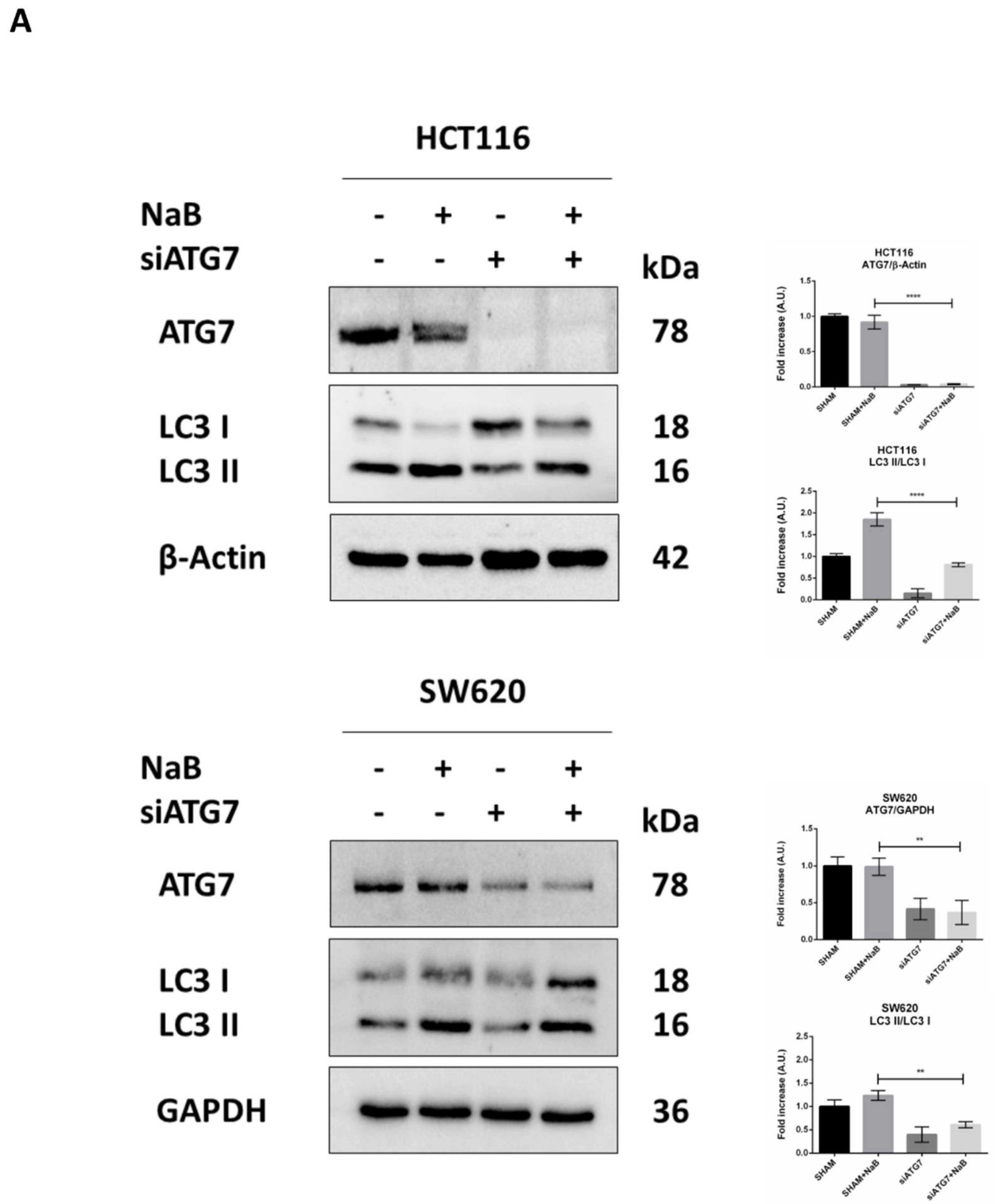
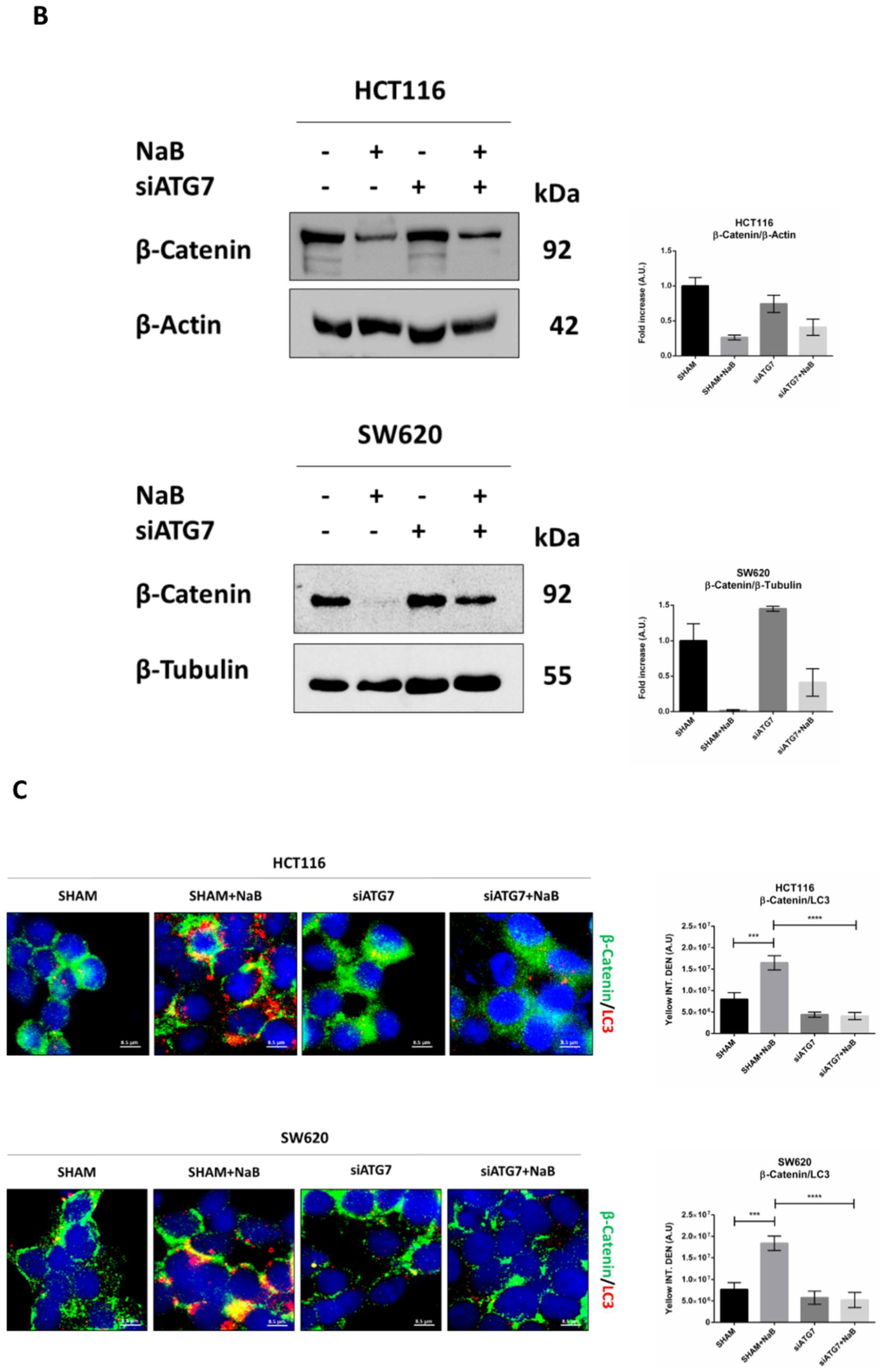
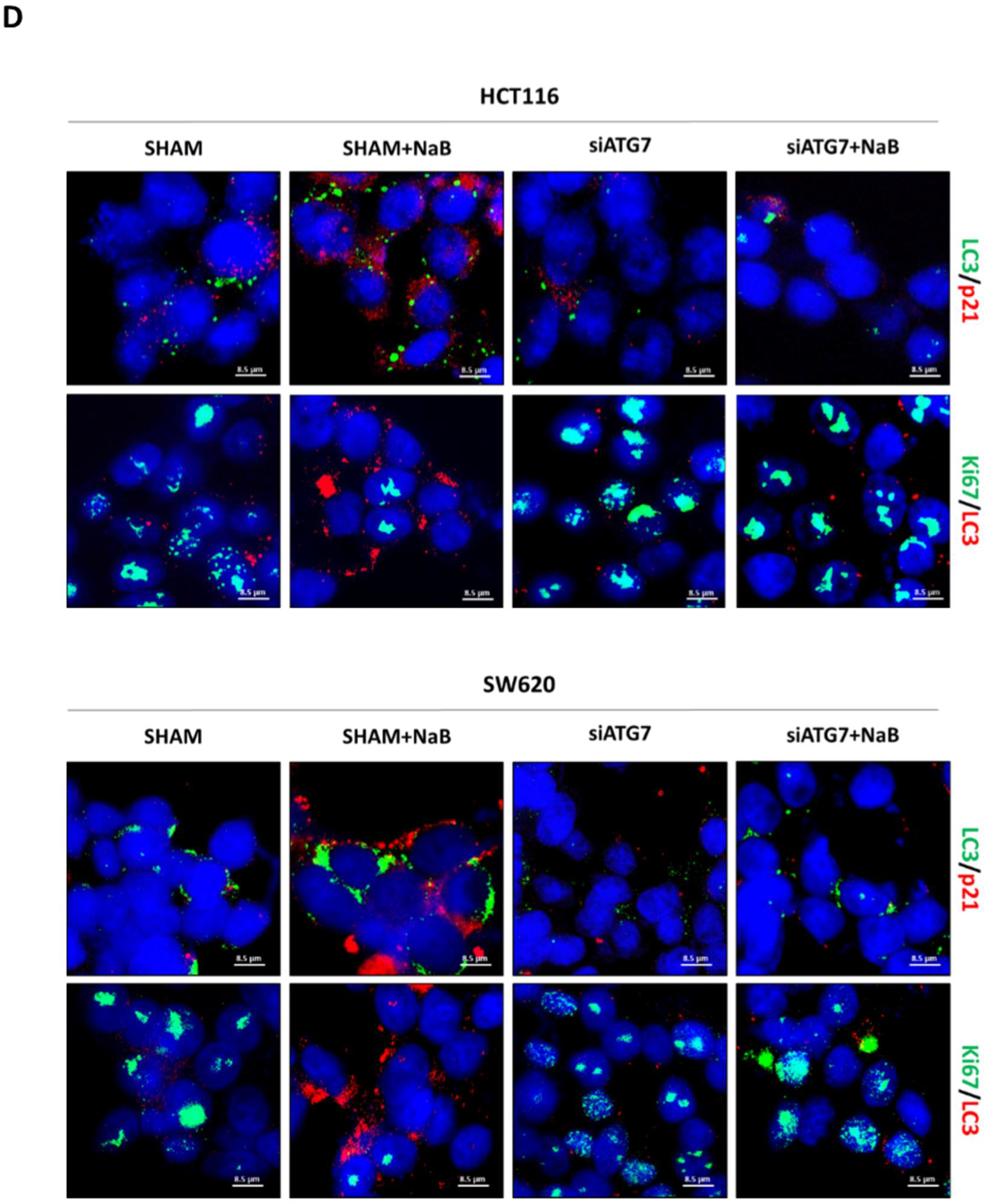
3.5. Disruption of Autophagy Prevents Butyrate-Induced β-Catenin Degradation and Inhibition of Cell Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of 494 Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Walker, L.C.; Robinson, B.A.; Frizelle, F.A.; Church, J.M.; Eglinton, T.W. Clinical implications of the genetics of sporadic colorectal cancer. ANZ J Surg. 2019, 89, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Mori, H. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci. 2007, 98, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Chen, J.; Deng, Z.L.; Luo, X.; Song, W.X.; Sharff, K.A.; Tang, N.; Haydon, R.C.; Luu, H.H.; He, T.C. Wnt signaling and human diseases: What are the therapeutic implications? Lab. Invest. 2007, 87, 97–103. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa EMelo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Liang, S.; Mao, Y.; Liao, M.; Xu, Y.; Chen, Y.; Huang, X.; Wei, C.; Wu, C.; Wang, Q.; Pan, X.; et al. Gut microbiome associated with APC gene mutation in patients with intestinal adenomatous polyps. Int. J. Biol. Sci. 2020, 16, 135–146. [Google Scholar] [CrossRef]
- Schneikert, J.; Behrens, J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007, 56, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat Rev Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Munemitsu, S.; Albert, I.; Rubinfeld, B.; Polakis, P. Deletion of an amino-terminal sequence beta-catenin in vivo and promotes hyperphosporylation of the adenomatous polyposis coli tumor suppressor protein. Mol. Cell Biol. 1996, 16, 4088–4094. [Google Scholar] [CrossRef] [Green Version]
- Inomata, M.; Ochiai, A.; Akimoto, S.; Kitano, S.; Hirohashi, S. Alteration of beta-catenin expression in colonic epithelial cells of familial adenomatous polyposis patients. Cancer Res. 1996, 56, 2213–2217. [Google Scholar]
- El-Bahrawy, M.; Poulsom, R.; Rowan, A.J.; Tomlinson, I.T.; Alison, M.R. Characterization of the E-cadherin/catenin complex in colorectal carcinoma cell lines. Int J Exp Pathol. 2004, 85, 65–74. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-Catenin-Tcf signaling in colon cancer by mutations in beta-Catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Gaujoux, S.; Launay, P.; Baudry, C.; Chokri, I.; Ragazzon, B.; Libé, R.; René-Corail, F.; Audebourg, A.; Vacher-Lavenu, M.C.; et al. Wnt/β-Catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: A study in cortisol-secreting and -nonsecreting tumors. J. Clin. Endocrinol. Metab. 2011, 96. [Google Scholar] [CrossRef] [Green Version]
- Azzopardi, D.; Dallosso, A.R.; Eliason, K.; Hendrickson, B.C.; Jones, N.; Rawstorne, E.; Colley, J.; Moskvina, V.; Frye, C.; Sampson, J.R.; et al. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 2008, 68, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Smits, R.; Kielman, M.F.; Breukel, C.; Zurcher, C.; Neufeld, K.; Jagmohan-Changur, S.; Hofland, N.; van Dijk, J.; White, R.; Edelmann, W.; et al. Apc1638T: A mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999, 13, 1309–1321. [Google Scholar] [CrossRef]
- Chen, D.; Jin, D.; Huang, S.; Wu, J.; Xu, M.; Liu, T.; Dong, W.; Liu, X.; Wang, S.; Zhong, W.; et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020, 469, 456–467. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Liu, Y.; Li, X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl. Oncol. 2020, 13, 100871. [Google Scholar] [CrossRef]
- Jin, L.; Chen, Y.; Cheng, D.; He, Z.; Shi, X.; Du, B.; Xi, X.; Gao, Y.; Guo, Y. YAP inhibits autophagy and promotes progression of colorectal cancer via upregulating Bcl-2 expression. Cell Death Dis. 2021, 12, 457. [Google Scholar] [CrossRef]
- Trincheri, N.F.; Follo, C.; Nicotra, G.; Peracchio, C.; Castino, R.; Isidoro, C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis 2008, 29, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Plasencia, C.; López-Urrutia, E.; García-Castillo, V.; Trujano-Camacho, S.; López-Camarillo, C.; Campos-Parra, A.D. Interplay Between Autophagy and Wnt/β-Catenin Signaling in Cancer: Therapeutic Potential Through Drug Repositioning. Front. Oncol. 2020, 10, 1037. [Google Scholar] [CrossRef]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef]
- Ferraresi, A.; Esposito, A.; Girone, C.; Vallino, L.; Salwa, A.; Ghezzi, I.; Thongchot, S.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts LPA-Induced Ovarian Cancer Cell Migration and Platinum Resistance by Rescuing Hedgehog-Mediated Autophagy. Cells 2021, 10, 3213. [Google Scholar] [CrossRef]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef] [Green Version]
- Salwa, A.; Ferraresi, A.; Chinthakindi, M.; Vallino, L.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. BECN1 and BRCA1 Deficiency Sensitizes Ovarian Cancer to Platinum Therapy and Confers Better Prognosis. Biomedicines 2021, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 3. [Google Scholar] [CrossRef]
- Shakoori, A.; Ougolkov, A.; Yu, Z.W.; Zhang, B.; Modarressi, M.H.; Billadeau, D.D.; Mai, M.; Takahashi, Y.; Minamoto, T. Deregulated GSK3beta activity in colorectal cancer: Its association with tumor cell survival and proliferation. Biochem. Biophys. Res. Commun. 2005, 334, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.F.; Awatade, N.T.; Loureiro, C.A.; Matos, P.; Amaral, M.D.; Jordan, P. The third dimension: New developments in cell culture models for colorectal research. Cell Mol. Life Sci. 2016, 73, 3971–3989. [Google Scholar] [CrossRef]
- Kumar, S.; Raina, K.; Agarwal, C.; Agarwal, R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell-enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget 2014, 5, 4972–4989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.N.; Choi, Y.S.; Kim, S.H.; Zhong, X.; Kim, W.; Park, J.S.; Saeidi, S.; Han, B.W.; Kim, N.; Lee, H.S.; et al. Resolvin D1 suppresses inflammation-associated tumorigenesis in the colon by inhibiting IL-6-induced mitotic spindle abnormality. FASEB J. 2021, 35, e21432. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Z.; Huang, Y.; Bai, C.; Zhang, X.; Fang, M.; Ju, Z.; Liu, B. Membrane dynamics of ATG4B and LC3 in autophagosome formation. J. Mol. Cell Biol. 2022, 13, 853–863. [Google Scholar] [CrossRef]
- Panebianco, C.; Potenza, A.; Andriulli, A.; Pazienza, V. Exploring the microbiota to better understand gastrointestinal cancers physiology. Clin. Chem. Lab. Med. 2018, 56, 1400–1412. [Google Scholar] [CrossRef]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.N.; Vitetta, L. Effects of Intestinal Microbial⁻Elaborated Butyrate on Oncogenic Signaling Pathways. Nutrients 2019, 11, 1026. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Yu, Y.; Wang, Y.Z.; Wang, J.J.; Guan, R.; Sun, Y.; Shi, F.; Gao, J.; Fu, X.L. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J. Cell Physiol. 2019, 234, 17023–17049. [Google Scholar] [CrossRef]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Hwang, J.H.; Choi, K.Y. Interaction of the Wnt/β-catenin and RAS-ERK pathways involving co-stabilization of both β-catenin and RAS plays important roles in the colorectal tumorigenesis. Adv. Biol. Regul. 2018, 68, 46–54. [Google Scholar] [CrossRef]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/β-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef]
- Polakis, P.; Hart, M.; Rubinfeld, B. Defects in the regulation of beta-Catenin in colorectal cancer. Adv. Exp. Med. Biol. 1999, 470, 23–32. [Google Scholar] [CrossRef]
- Thongchot, S.; Ferraresi, A.; Vidoni, C.; Loilome, W.; Yongvanit, P.; Namwat, N.; Isidoro, C. Resveratrol interrupts the pro-invasive communication between Cancer associated Fibroblasts and Cholangiocarcinoma cells. Cancer Lett. 2018, 434, 206–207. [Google Scholar] [CrossRef]
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients--a summary of published results. Int. J. Colorectal. Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer Cells. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6-a key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRAF | KRAS | TP53 | PTEN | APC | CTNNB1 | |
|---|---|---|---|---|---|---|
| HCT116 | I666I Silent P301P Silent | G13D (Missense) | Wild-type | Wild-type | Wild-type | S45del |
| SW620 | Wild-type | G12V | R273H P309S | Wild-type | Q1338 (Nonsense) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garavaglia, B.; Vallino, L.; Ferraresi, A.; Esposito, A.; Salwa, A.; Vidoni, C.; Gentilli, S.; Isidoro, C. Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status. Biomedicines 2022, 10, 1131. https://doi.org/10.3390/biomedicines10051131
Garavaglia B, Vallino L, Ferraresi A, Esposito A, Salwa A, Vidoni C, Gentilli S, Isidoro C. Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status. Biomedicines. 2022; 10(5):1131. https://doi.org/10.3390/biomedicines10051131
Chicago/Turabian StyleGaravaglia, Beatrice, Letizia Vallino, Alessandra Ferraresi, Andrea Esposito, Amreen Salwa, Chiara Vidoni, Sergio Gentilli, and Ciro Isidoro. 2022. "Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status" Biomedicines 10, no. 5: 1131. https://doi.org/10.3390/biomedicines10051131
APA StyleGaravaglia, B., Vallino, L., Ferraresi, A., Esposito, A., Salwa, A., Vidoni, C., Gentilli, S., & Isidoro, C. (2022). Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status. Biomedicines, 10(5), 1131. https://doi.org/10.3390/biomedicines10051131