
The Impact of RIPK1 Kinase Inhibition on Atherogenesis: A Genetic and a Pharmacological Approach

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Echocardiography
2.3. Histological Analyses
2.4. Cell Culture
2.5. Western Blotting
2.6. Statistical Analyses
3. Results
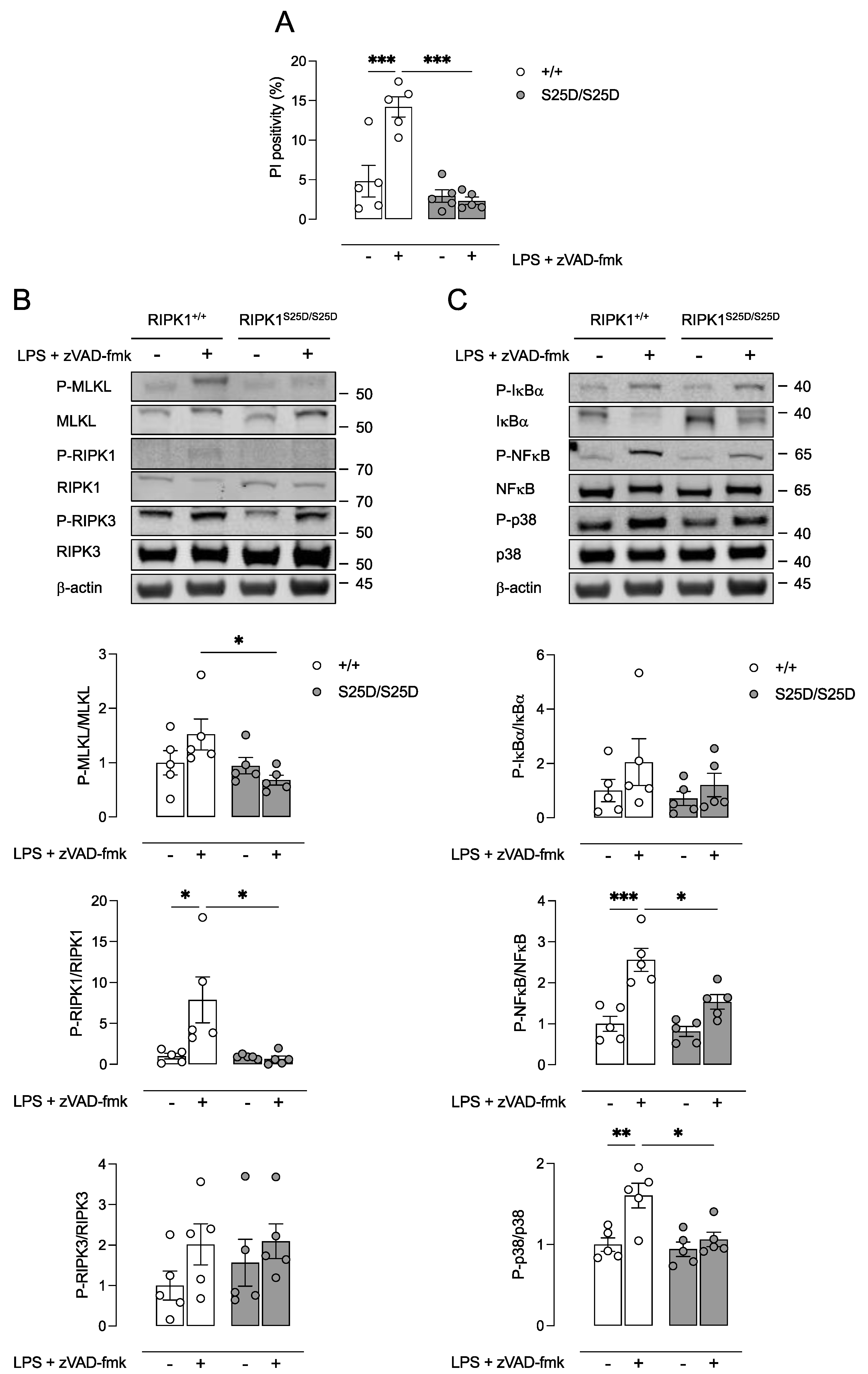
3.1. RIPK1S25D/S25D BMDMs Are Protected against Necroptosis
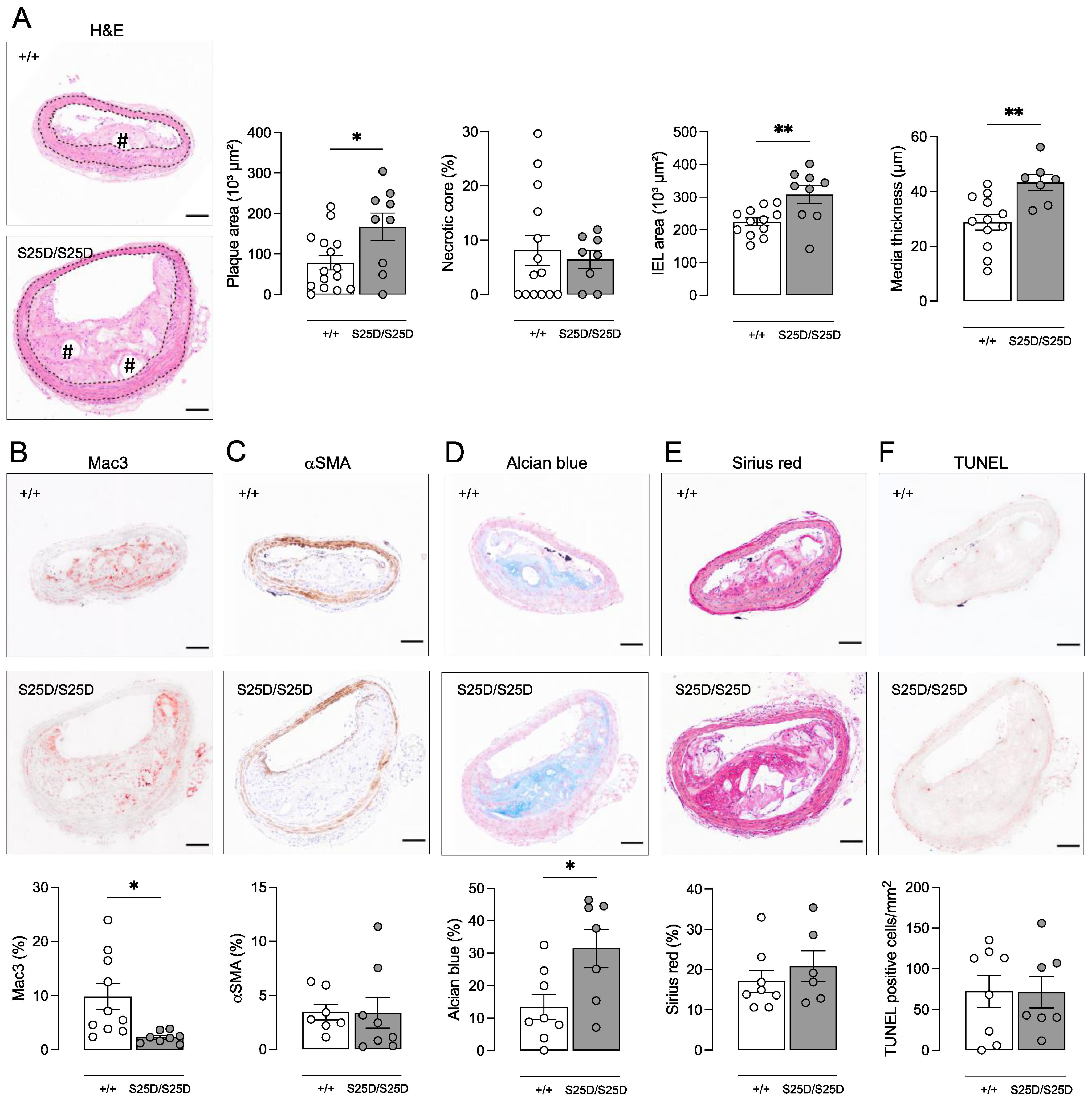
3.2. ApoE−/− RIPK1S25D/S25D Mice Develop Larger Plaques with Increased Deposition of Extracellular Matrix Components
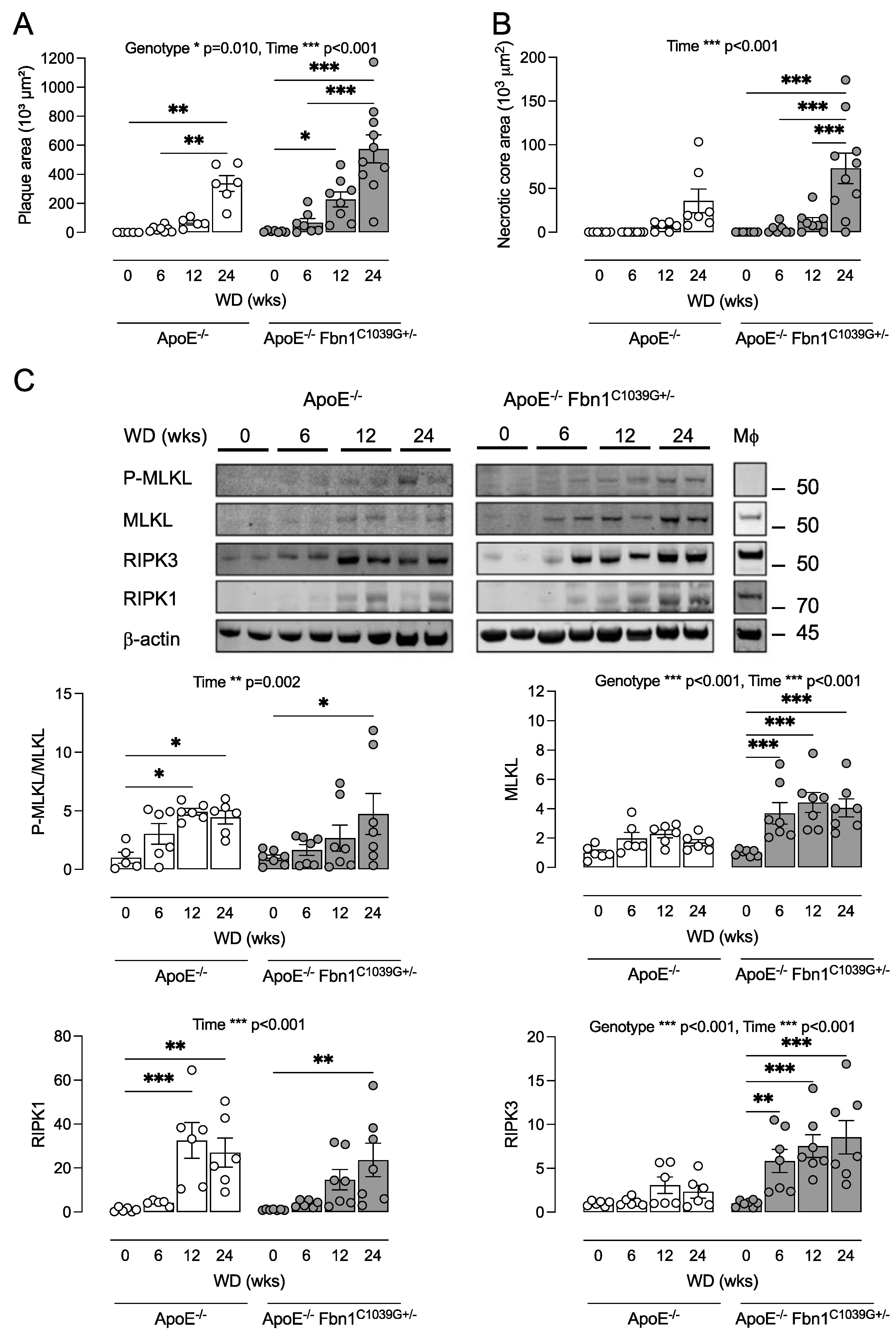
3.3. ApoE−/− Fbn1C1039G+/− Mice Can Be Used as a Tool to Study Necroptosis in Atherosclerosis
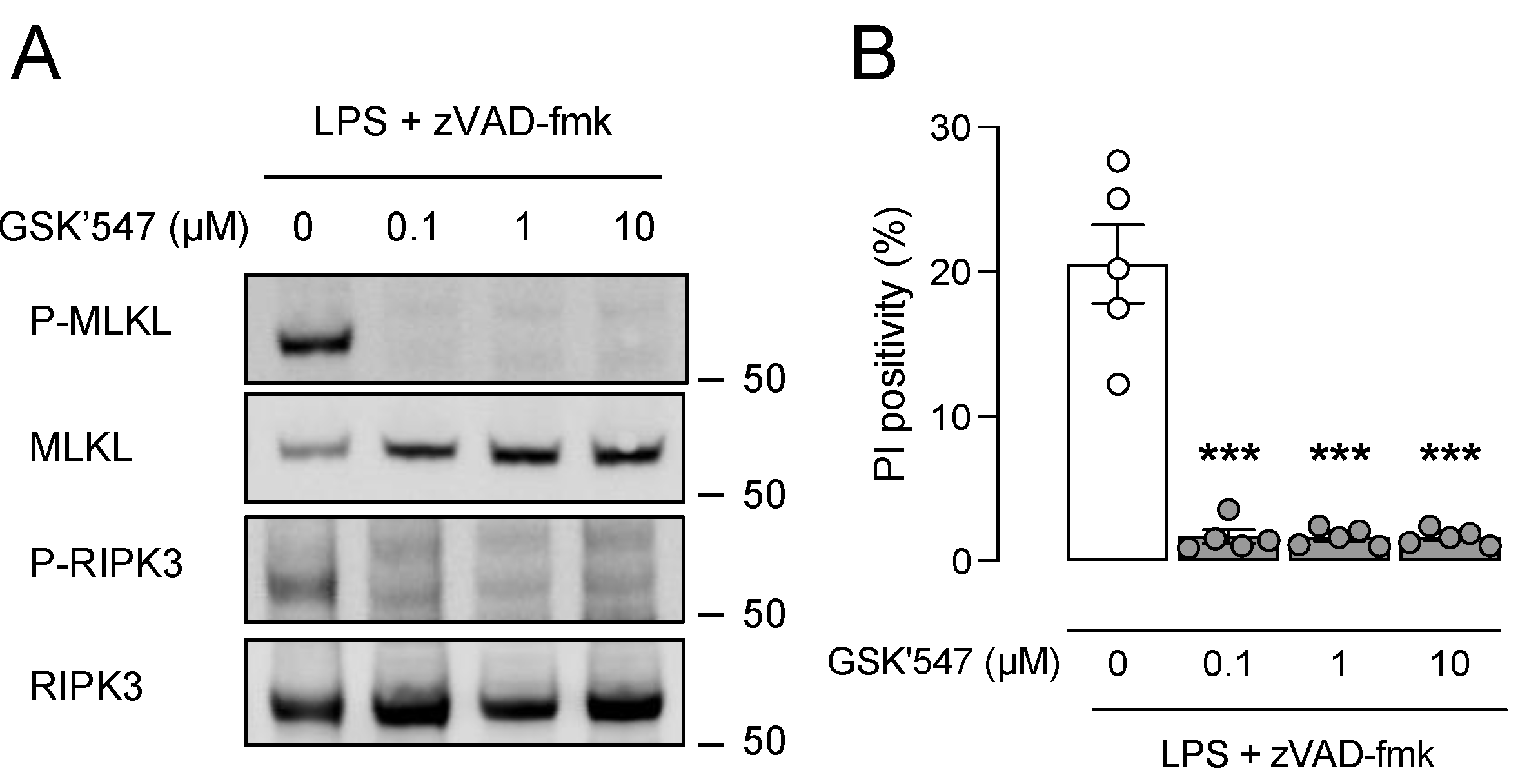
3.4. Pharmacological Inhibition of RIPK1 with GSK’547 Does Not Alter Plaque Size and Composition in ApoE−/− Fbn1C1039G+/− Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Fact Sheet Cardiovascular Diseases; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the Vulnerable Plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A Role of RIP3-Mediated Macrophage Necrosis in Atherosclerosis Development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, A.; Robichaud, S.; Nguyen, M.-A.; Geoffrion, M.; Wyatt, H.; Cottee, M.L.; Dennison, T.; Pietrangelo, A.; Lee, R.; Lagace, T.A.; et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) Decreases Necrotic Core but Increases Macrophage Lipid Accumulation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhang, Y.; Kuang, Y.; Ma, Q. Changes in plasma levels of ripk1, ripk3, and mlkl in patients with coronary atherosclerotic heart disease and its clinical predictive value. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2020, 45, 1096–1103. [Google Scholar] [PubMed]
- Coornaert, I.; Puylaert, P.; Marcasolli, G.; Grootaert, M.O.; Vandenabeele, P.; De Meyer, G.R.; Martinet, W. Impact of myeloid RIPK1 gene deletion on atherogenesis in ApoE-deficient mice. Atherosclerosis 2021, 322, 51–60. [Google Scholar] [CrossRef]
- Karunakaran, D.; Nguyen, M.-A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates with Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce nf-κb Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef]
- Meng, L.; Jin, W.; Wang, X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc. Natl. Acad. Sci. USA 2015, 112, 11007–11012. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, F.; Wang, X. TNF-α Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Delanghe, T.; Dondelinger, Y.; Bertrand, M.J. RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol. 2020, 30, 189–200. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Jouan, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. Nf-kappab-Independent Role of ikkalpha/ikkbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase rip mediates the tnf-induced nf-kappab signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colijn, S.; Muthukumar, V.; Xie, J.; Gao, S.; Griffin, C.T. Cell-specific and athero-protective roles for RIPK3 in a murine model of atherosclerosis. Dis. Models Mech. 2020, 13, dmm041962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Herck, J.L.; De Meyer, G.R.; Martinet, W.; Van Hove, C.E.; Foubert, K.; Theunis, M.H.; Apers, S.; Bult, H.; Vrints, C.J.; Herman, A.G. Impaired Fibrillin-1 Function Promotes Features of Plaque Instability in Apolipoprotein E–Deficient Mice. Circulation 2009, 120, 2478–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Donckt, C.; Van Herck, J.L.; Schrijvers, D.M.; Vanhoutte, G.; Verhoye, M.; Blockx, I.; Van Der Linden, A.; Bauters, D.; Lijnen, H.R.; Sluimer, J.C.; et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur. Heart J. 2015, 36, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, G.; Nicoletti, A.; Zhou, X.; Törnberg, I.; Hansson, G.K. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis 1999, 145, 301–308. [Google Scholar] [CrossRef]
- Mansukhani, N.A.; Wang, Z.; Shively, V.P.; Kelly, M.E.; Vercammen, J.M.; Kibbe, M.R. Sex differences in the LDL receptor knockout mouse model of atherosclerosis. Artery Res. 2017, 20, 8–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Huang, Y.; Chen, H.; Rao, H.; Yang, G.; Wan, Q.; Peng, Z.; Bertin, J.; Geddes, B.; et al. Stage-Dependent Impact of RIPK1 Inhibition on Atherogenesis: Dual Effects on Inflammation and Foam Cell Dynamics. Front. Cardiovasc. Med. 2021, 8, 715337. [Google Scholar] [CrossRef]
- Kurdi, A.; Roth, L.; Van der Veken, B.; Van Dam, D.; De Deyn, P.P.; De Doncker, M.; Neels, H.; De Meyer, G.R.; Martinet, W. Everolimus depletes plaque macrophages, abolishes intraplaque neovascularization and improves survival in mice with advanced atherosclerosis. Vasc. Pharmacol. 2019, 113, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lascio, N.; Stea, F.; Kusmic, C.; Sicari, R.; Faita, F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014, 237, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Johnson, J.; Carson, K.G.S.; Jackson, C.L. Characteristics of Intact and Ruptured Atherosclerotic Plaques in Brachiocephalic Arteries of Apolipoprotein E Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengrenyuk, Y.; Kaplan, T.J.; Cardoso, L.; Randolph, G.J.; Weinbaum, S. Computational stress analysis of atherosclerotic plaques in ApoE knockout mice. Ann. Biomed. Eng. 2010, 38, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Glagov, S.; Weisenberg, E.; Zarins, C.K.; Stankunavicius, R.; Kolettis, G.J. Compensatory Enlargement of Human Atherosclerotic Coronary Arteries. N. Engl. J. Med. 1987, 316, 1371–1375. [Google Scholar] [CrossRef]
- Bonthu, S.; Heistad, D.D.; Chappell, D.A.; Lamping, K.G.; Faraci, F. Atherosclerosis, Vascular Remodeling, and Impairment of Endothelium-Dependent Relaxation in Genetically Altered Hyperlipidemic Mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2333–2340. [Google Scholar] [CrossRef]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef] [Green Version]
- Roth, L.; Rombouts, M.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R. Cholesterol-independent effects of atorvastatin prevent cardiovascular morbidity and mortality in a mouse model of atherosclerotic plaque rupture. Vasc. Pharmacol. 2016, 80, 50–58. [Google Scholar] [CrossRef]
- Wang, L.; Chang, X.; Feng, J.; Yu, J.; Chen, G. TRADD Mediates RIPK1-Independent Necroptosis Induced by Tumor Necrosis Factor. Front. Cell Dev. Biol. 2020, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.-D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Schrijvers, D.; De Meyer, G. Necrotic cell death in atherosclerosis. Basic Res. Cardiol. 2011, 106, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, D.M.; De Meyer, G.R.; Kockx, M.M.; Herman, A.G.; Martinet, W. Phagocytosis of Apoptotic Cells by Macrophages Is Impaired in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Aterioscler. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puylaert, P.; Coornaert, I.; Neutel, C.H.G.; Dondelinger, Y.; Delanghe, T.; Bertrand, M.J.M.; Guns, P.-J.; De Meyer, G.R.Y.; Martinet, W. The Impact of RIPK1 Kinase Inhibition on Atherogenesis: A Genetic and a Pharmacological Approach. Biomedicines 2022, 10, 1016. https://doi.org/10.3390/biomedicines10051016
Puylaert P, Coornaert I, Neutel CHG, Dondelinger Y, Delanghe T, Bertrand MJM, Guns P-J, De Meyer GRY, Martinet W. The Impact of RIPK1 Kinase Inhibition on Atherogenesis: A Genetic and a Pharmacological Approach. Biomedicines. 2022; 10(5):1016. https://doi.org/10.3390/biomedicines10051016
Chicago/Turabian StylePuylaert, Pauline, Isabelle Coornaert, Cédric H. G. Neutel, Yves Dondelinger, Tom Delanghe, Mathieu J. M. Bertrand, Pieter-Jan Guns, Guido R. Y. De Meyer, and Wim Martinet. 2022. "The Impact of RIPK1 Kinase Inhibition on Atherogenesis: A Genetic and a Pharmacological Approach" Biomedicines 10, no. 5: 1016. https://doi.org/10.3390/biomedicines10051016
APA StylePuylaert, P., Coornaert, I., Neutel, C. H. G., Dondelinger, Y., Delanghe, T., Bertrand, M. J. M., Guns, P.-J., De Meyer, G. R. Y., & Martinet, W. (2022). The Impact of RIPK1 Kinase Inhibition on Atherogenesis: A Genetic and a Pharmacological Approach. Biomedicines, 10(5), 1016. https://doi.org/10.3390/biomedicines10051016