
Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor

 , , ,
, , ,
Abstract
:1. Introduction
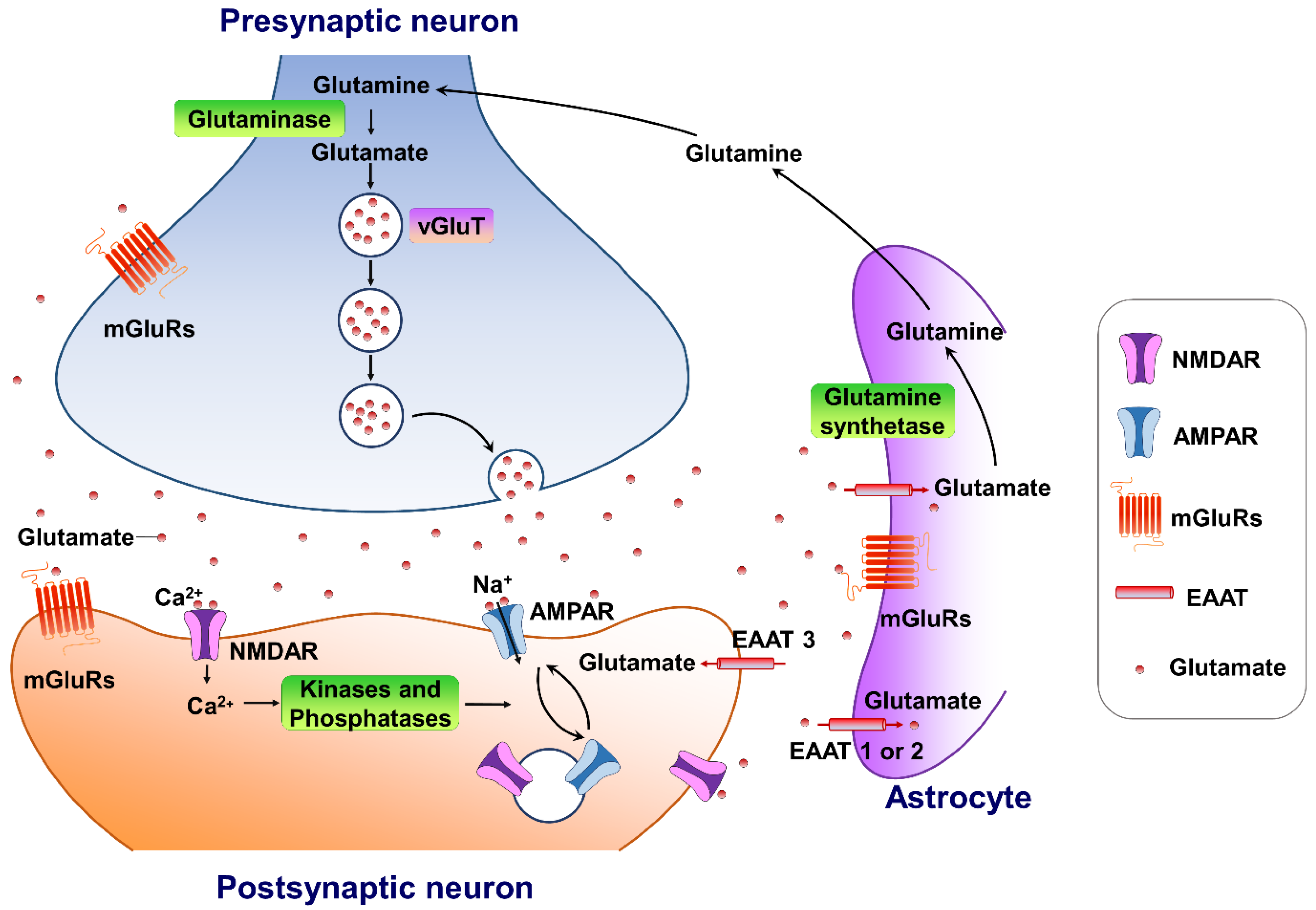
2. A Brief Overview of Glutamate Neurotransmission in the Brain
3. Synaptic Plasticity of Chronic Stressor Priming
3.1. Hippocampus
3.2. Amygdala
3.3. Prefrontal Cortex
3.4. Nucleus Accumbens
3.5. Periaqueductal Gray
4. Targeting AMPA-Glutamatergic Signaling for the Development of Novel Therapeutics for Depressive Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, B.; Conde, T.; Domingues, I.; Domingues, M.R. Adaptation of Lipid Profiling in Depression Disease and Treatment: A Critical Review. Int. J. Mol. Sci. 2022, 23, 2032. [Google Scholar] [CrossRef]
- Belmaker, R.H.; Agam, G. Major depressive disorder. N. Engl. J. Med. 2008, 358, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fife, D.; Wang, G.; Sheehan, J.J.; Boden, R.; Brandt, L.; Brenner, P.; Reutfors, J.; DiBernardo, A. All-cause mortality in patients with treatment-resistant depression: A cohort study in the US population. Ann. Gen. Psychiatry 2019, 18, 23. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.C.; Jacob, S.A.; Tangiisuran, B. Barriers and facilitators of adherence to antidepressants among outpatients with major depressive disorder: A qualitative study. PLoS ONE 2017, 12, e0179290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.I. Factors predicting adherence to antidepressant treatment. Curr. Opin. Psychiatry 2014, 27, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Jesulola, E.; Micalos, P.; Baguley, I.J. Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model—Are we there yet? Behav. Brain Res. 2018, 341, 79–90. [Google Scholar] [CrossRef]
- Manji, H.K.; Drevets, W.C.; Charney, D.S. The cellular neurobiology of depression. Nat. Med. 2001, 7, 541–547. [Google Scholar] [CrossRef]
- Heninger, G.R.; Delgado, P.L.; Charney, D.S. The revised monoamine theory of depression: A modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry 1996, 29, 2–11. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6. [Google Scholar]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Iob, E.; Kirschbaum, C.; Steptoe, A. Persistent depressive symptoms, HPA-axis hyperactivity, and inflammation: The role of cognitive-affective and somatic symptoms. Mol. Psychiatry 2020, 25, 1130–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, C.; Newport, D.J.; Wagner, D.; Wilcox, M.M.; Miller, A.H.; Nemeroff, C.B. The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: A multiple regression analysis. Depress. Anxiety 2002, 15, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Skorzewska, A.; Lehner, M.; Wislowska-Stanek, A.; Krzascik, P.; Ziemba, A.; Plaznik, A. The effect of chronic administration of corticosterone on anxiety- and depression-like behavior and the expression of GABA-A receptor alpha-2 subunits in brain structures of low- and high-anxiety rats. Horm. Behav. 2014, 65, 6–13. [Google Scholar] [CrossRef]
- Kim, J.G.; Jung, H.S.; Kim, K.J.; Min, S.S.; Yoon, B.J. Basal blood corticosterone level is correlated with susceptibility to chronic restraint stress in mice. Neurosci. Lett. 2013, 555, 137–142. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef]
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.P.; Thaller, C.; Zoghbi, H.Y. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef] [Green Version]
- Nandam, L.S.; Brazel, M.; Zhou, M.; Jhaveri, D.J. Cortisol and Major Depressive Disorder-Translating Findings From Humans to Animal Models and Back. Front. Psychiatry 2019, 10, 974. [Google Scholar] [CrossRef]
- Faravelli, C.; Lo Sauro, C.; Godini, L.; Lelli, L.; Benni, L.; Pietrini, F.; Lazzeretti, L.; Talamba, G.A.; Fioravanti, G.; Ricca, V. Childhood stressful events, HPA axis and anxiety disorders. World J. Psychiatry 2012, 2, 13–25. [Google Scholar] [CrossRef]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M., Jr.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E., Jr.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658. [Google Scholar] [CrossRef] [Green Version]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Freed, W.J.; Dillon-Carter, O.; Kleinman, J.E. Properties of [3H]AMPA binding in postmortem human brain from psychotic subjects and controls: Increases in caudate nucleus associated with suicide. Exp. Neurol. 1993, 121, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Meador-Woodruff, J.H.; Hogg, A.J., Jr.; Smith, R.E. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res. Bull. 2001, 55, 631–640. [Google Scholar] [CrossRef]
- Zarate, C.A.; Quiroz, J.; Payne, J.; Manji, H.K. Modulators of the glutamatergic system: Implications for the development of improved therapeutics in mood disorders. Psychopharmacol. Bull. 2002, 36, 35–83. [Google Scholar]
- Takamori, S.; Rhee, J.S.; Rosenmund, C.; Jahn, R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 2000, 407, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Watkins, J.C.; Jane, D.E. The glutamate story. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S100–S108. [Google Scholar] [CrossRef]
- Sheline, Y.I.; Liston, C.; McEwen, B.S. Parsing the Hippocampus in Depression: Chronic Stress, Hippocampal Volume, and Major Depressive Disorder. Biol. Psychiatry 2019, 85, 436–438. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Campbell, S.; McEwen, B.S.; Macdonald, K.; Amano, S.; Joffe, R.T.; Nahmias, C.; Young, L.T. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc. Natl. Acad. Sci. USA 2003, 100, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef] [PubMed]
- Schloesser, R.J.; Manji, H.K.; Martinowich, K. Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport 2009, 20, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, N.; Lukoyanov, N.V.; Madeira, M.D.; Almeida, O.F.; Paula-Barbosa, M.M. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 2000, 97, 253–266. [Google Scholar] [CrossRef]
- Magarinos, A.M.; Li, C.J.; Gal Toth, J.; Bath, K.G.; Jing, D.; Lee, F.S.; McEwen, B.S. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus 2011, 21, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Lee, H.; Young, A.H.; Aarsland, D.; Thuret, S. Adult Hippocampal Neurogenesis in Major Depressive Disorder and Alzheimer’s Disease. Trends Mol. Med. 2020, 26, 803–818. [Google Scholar] [CrossRef]
- Soetanto, A.; Wilson, R.S.; Talbot, K.; Un, A.; Schneider, J.A.; Sobiesk, M.; Kelly, J.; Leurgans, S.; Bennett, D.A.; Arnold, S.E. Association of anxiety and depression with microtubule-associated protein 2- and synaptopodin-immunolabeled dendrite and spine densities in hippocampal CA3 of older humans. Arch. Gen. Psychiatry 2010, 67, 448–457. [Google Scholar] [CrossRef]
- Ma, H.; Li, C.; Wang, J.; Zhang, X.; Li, M.; Zhang, R.; Huang, Z.; Zhang, Y. Amygdala-hippocampal innervation modulates stress-induced depressive-like behaviors through AMPA receptors. Proc. Natl. Acad. Sci. USA 2021, 118, e2019409118. [Google Scholar] [CrossRef]
- Calfa, G.; Bussolino, D.; Molina, V.A. Involvement of the lateral septum and the ventral Hippocampus in the emotional sequelae induced by social defeat: Role of glucocorticoid receptors. Behav. Brain Res. 2007, 181, 23–34. [Google Scholar] [CrossRef]
- Pittman, Q.J.; Blume, H.W.; Renaud, L.P. Connections of the hypothalamic paraventricular nucleus with the neurohypophysis, median eminence, amygdala, lateral septum and midbrain periaqueductal gray: An electrophysiological study in the rat. Brain Res. 1981, 215, 15–28. [Google Scholar] [CrossRef]
- Radley, J.J.; Anderson, R.M.; Hamilton, B.A.; Alcock, J.A.; Romig-Martin, S.A. Chronic stress-induced alterations of dendritic spine subtypes predict functional decrements in an hypothalamo-pituitary-adrenal-inhibitory prefrontal circuit. J. Neurosci. 2013, 33, 14379–14391. [Google Scholar] [CrossRef] [Green Version]
- Karst, H.; Joels, M. Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J. Neurophysiol. 2005, 94, 3479–3486. [Google Scholar] [CrossRef]
- Kallarackal, A.J.; Kvarta, M.D.; Cammarata, E.; Jaberi, L.; Cai, X.; Bailey, A.M.; Thompson, S.M. Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J. Neurosci. 2013, 33, 15669–15674. [Google Scholar] [CrossRef] [Green Version]
- Kvarta, M.D.; Bradbrook, K.E.; Dantrassy, H.M.; Bailey, A.M.; Thompson, S.M. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol. 2015, 114, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schutz, G.; Joels, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Zheng, H.L.; Luo, Y.; He, J.G.; Wang, W.; Han, J.; Zhang, L.; Wang, X.; Ni, L.; Zhou, H.Y.; et al. Gene deficiency and pharmacological inhibition of caspase-1 confers resilience to chronic social defeat stress via regulating the stability of surface AMPARs. Mol. Psychiatry 2018, 23, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Sharp, B.M. Basolateral amygdala and stress-induced hyperexcitability affect motivated behaviors and addiction. Transl. Psychiatry 2017, 7, e1194. [Google Scholar] [CrossRef]
- Zhou, H.Y.; He, J.G.; Hu, Z.L.; Xue, S.G.; Xu, J.F.; Cui, Q.Q.; Gao, S.Q.; Zhou, B.; Wu, P.F.; Long, L.H.; et al. A-Kinase Anchoring Protein 150 and Protein Kinase A Complex in the Basolateral Amygdala Contributes to Depressive-like Behaviors Induced by Chronic Restraint Stress. Biol. Psychiatry 2019, 86, 131–142. [Google Scholar] [CrossRef]
- Yi, E.S.; Oh, S.; Lee, J.K.; Leem, Y.H. Chronic stress-induced dendritic reorganization and abundance of synaptosomal PKA-dependent CP-AMPA receptor in the basolateral amygdala in a mouse model of depression. Biochem. Biophys. Res. Commun. 2017, 486, 671–678. [Google Scholar] [CrossRef]
- Hubert, G.W.; Li, C.; Rainnie, D.G.; Muly, E.C. Effects of stress on AMPA receptor distribution and function in the basolateral amygdala. Brain Struct. Funct. 2014, 219, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Padival, M.; Quinette, D.; Rosenkranz, J.A. Effects of repeated stress on excitatory drive of basal amygdala neurons in vivo. Neuropsychopharmacology 2013, 38, 1748–1762. [Google Scholar] [CrossRef] [Green Version]
- Kuniishi, H.; Yamada, D.; Wada, K.; Yamada, M.; Sekiguchi, M. Stress induces insertion of calcium-permeable AMPA receptors in the OFC-BLA synapse and modulates emotional behaviours in mice. Transl. Psychiatry 2020, 10, 154. [Google Scholar] [CrossRef]
- Phelps, E.A.; LeDoux, J.E. Contributions of the amygdala to emotion processing: From animal models to human behavior. Neuron 2005, 48, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Koenigs, M.; Grafman, J. The functional neuroanatomy of depression: Distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav. Brain Res. 2009, 201, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Pantazatos, S.P.; Huang, Y.Y.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; Mann, J.J. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: Evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatry 2017, 22, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Woo, E.; Sansing, L.H.; Arnsten, A.F.T.; Datta, D. Chronic Stress Weakens Connectivity in the Prefrontal Cortex: Architectural and Molecular Changes. Chronic Stress 2021, 5, 24705470211029254. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Liu, W.; Karatsoreos, I.N.; Ren, Y.; Feng, J.; McEwen, B.S.; Yan, Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol. Psychiatry 2011, 16, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Zhong, P.; Qin, L.; Tan, T.; Yan, Z. Chemicogenetic Restoration of the Prefrontal Cortex to Amygdala Pathway Ameliorates Stress-Induced Deficits. Cereb. Cortex 2018, 28, 1980–1990. [Google Scholar] [CrossRef]
- Seo, J.S.; Wei, J.; Qin, L.; Kim, Y.; Yan, Z.; Greengard, P. Cellular and molecular basis for stress-induced depression. Mol. Psychiatry 2017, 22, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Xiong, Z.; Lee, J.B.; Cheng, J.; Duffney, L.J.; Matas, E.; Yan, Z. Histone Modification of Nedd4 Ubiquitin Ligase Controls the Loss of AMPA Receptors and Cognitive Impairment Induced by Repeated Stress. J. Neurosci. 2016, 36, 2119–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, M.; Gruca, P.; Lason, M.; Litwa, E.; Solecki, W.; Willner, P. AMPA receptors mediate the pro-cognitive effects of electrical and optogenetic stimulation of the medial prefrontal cortex in antidepressant non-responsive Wistar-Kyoto rats. J. Psychopharmacol. 2020, 34, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Sesack, S.R.; Grace, A.A. Cortico-Basal Ganglia reward network: Microcircuitry. Neuropsychopharmacology 2010, 35, 27–47. [Google Scholar] [CrossRef]
- Heshmati, M.; Russo, S.J. Anhedonia and the brain reward circuitry in depression. Curr. Behav. Neurosci. Rep. 2015, 2, 146–153. [Google Scholar] [CrossRef]
- Smith, K.S.; Berridge, K.C.; Aldridge, J.W. Disentangling pleasure from incentive salience and learning signals in brain reward circuitry. Proc. Natl. Acad. Sci. USA 2011, 108, E255–E264. [Google Scholar] [CrossRef] [Green Version]
- Bessa, J.M.; Morais, M.; Marques, F.; Pinto, L.; Palha, J.A.; Almeida, O.F.; Sousa, N. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl. Psychiatry 2013, 3, e266. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wang, Y.; Chen, X.; Zhang, Z.; Xiao, L.; Zhou, Y. Anhedonia correlates with functional connectivity of the nucleus accumbens subregions in patients with major depressive disorder. Neuroimage Clin. 2021, 30, 102599. [Google Scholar] [CrossRef]
- Lim, B.K.; Huang, K.W.; Grueter, B.A.; Rothwell, P.E.; Malenka, R.C. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 2012, 487, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Bandler, R.; Keay, K.A.; Floyd, N.; Price, J. Central circuits mediating patterned autonomic activity during active vs. passive emotional coping. Brain Res. Bull. 2000, 53, 95–104. [Google Scholar] [CrossRef]
- Ho, Y.C.; Lee, H.J.; Tung, L.W.; Liao, Y.Y.; Fu, S.Y.; Teng, S.F.; Liao, H.T.; Mackie, K.; Chiou, L.C. Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. J. Neurosci. 2011, 31, 14600–14610. [Google Scholar] [CrossRef]
- Ho, Y.C.; Cheng, J.K.; Chiou, L.C. Impairment of adenylyl cyclase-mediated glutamatergic synaptic plasticity in the periaqueductal grey in a rat model of neuropathic pain. J. Physiol. 2015, 593, 2955–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.H.; Kan, H.W.; Ho, Y.C. Periaqueductal gray is required for controlling chronic stress-induced depression-like behavior. Biochem. Biophys. Res. Commun. 2022, 593, 28–34. [Google Scholar] [CrossRef]
- Ho, Y.C.; Cheng, J.K.; Chiou, L.C. Hypofunction of glutamatergic neurotransmission in the periaqueductal gray contributes to nerve-injury-induced neuropathic pain. J. Neurosci. 2013, 33, 7825–7836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.T.; Chen, Y.H.; Mackie, K.; Chiou, L.C. Median Nerve Stimulation as a Nonpharmacological Approach to Bypass Analgesic Tolerance to Morphine: A Proof-of-Concept Study in Mice. J. Pain. 2021, 22, 300–312. [Google Scholar] [CrossRef]
- Lee, M.T.; Chiu, Y.T.; Chiu, Y.C.; Hor, C.C.; Lee, H.J.; Guerrini, R.; Calo, G.; Chiou, L.C. Neuropeptide S-initiated sequential cascade mediated by OX1, NK1, mGlu5 and CB1 receptors: A pivotal role in stress-induced analgesia. J. Biomed. Sci. 2020, 27, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.T.; Mackie, K.; Chiou, L.C. Alternative pain management via endocannabinoids in the time of the opioid epidemic: Peripheral neuromodulation and pharmacological interventions. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Yang, P.S.; Peng, H.Y.; Lin, T.B.; Hsieh, M.C.; Lai, C.Y.; Lee, A.S.; Wang, H.H.; Ho, Y.C. NMDA receptor partial agonist GLYX-13 alleviates chronic stress-induced depression-like behavior through enhancement of AMPA receptor function in the periaqueductal gray. Neuropharmacology 2020, 178, 108269. [Google Scholar] [CrossRef]
- Chou, D.; Peng, H.Y.; Lin, T.B.; Lai, C.Y.; Hsieh, M.C.; Wen, Y.C.; Lee, A.S.; Wang, H.H.; Yang, P.S.; Chen, G.D.; et al. (2R,6R)-hydroxynorketamine rescues chronic stress-induced depression-like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology 2018, 139, 1–12. [Google Scholar] [CrossRef]
- Ho, Y.C.; Lin, T.B.; Hsieh, M.C.; Lai, C.Y.; Chou, D.; Chau, Y.P.; Chen, G.D.; Peng, H.Y. Periaqueductal Gray Glutamatergic Transmission Governs Chronic Stress-Induced Depression. Neuropsychopharmacology 2018, 43, 302–312. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Liu, B.; Li, C.; Liu, Z.; Deng, J.; Luo, H.; Li, X.; Wu, J.; Li, H.; et al. Involvement of Scratch2 in GalR1-mediated depression-like behaviors in the rat ventral periaqueductal gray. Proc. Natl. Acad. Sci. USA 2021, 118, e1922586118. [Google Scholar] [CrossRef]
- Wang, P.; Li, H.; Barde, S.; Zhang, M.D.; Sun, J.; Wang, T.; Zhang, P.; Luo, H.; Wang, Y.; Yang, Y.; et al. Depression-like behavior in rat: Involvement of galanin receptor subtype 1 in the ventral periaqueductal gray. Proc. Natl. Acad. Sci. USA 2016, 113, E4726–E4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskal, J.R.; Burgdorf, J.S.; Stanton, P.K.; Kroes, R.A.; Disterhoft, J.F.; Burch, R.M.; Khan, M.A. The Development of Rapastinel (Formerly GLYX-13); A Rapid Acting and Long Lasting Antidepressant. Curr. Neuropharmacol. 2017, 15, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Fogaca, M.V.; Deyama, S.; Li, X.Y.; Fukumoto, K.; Duman, R.S. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry 2018, 23, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Duman, C.; Kato, T.; Hare, B.; Lopresto, D.; Bang, E.; Burgdorf, J.; Moskal, J.; Taylor, J.; Aghajanian, G.; et al. GLYX-13 Produces Rapid Antidepressant Responses with Key Synaptic and Behavioral Effects Distinct from Ketamine. Neuropsychopharmacology 2017, 42, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Lavialle, M.; Aumann, G.; Anlauf, E.; Prols, F.; Arpin, M.; Derouiche, A. Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 12915–12919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Wang, Z.; Wang, H.; Li, H.; Huang, C.; Shen, Y.; Ma, X.; Sun, H. Neurons and Astrocytes in Ventrolateral Periaqueductal Gray Contribute to Restraint Water Immersion Stress-Induced Gastric Mucosal Damage via the ERK1/2 Signaling Pathway. Int. J. Neuropsychopharmacol. 2021, 24, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Imbe, H.; Kimura, A.; Donishi, T.; Kaneoke, Y. Chronic restraint stress decreases glial fibrillary acidic protein and glutamate transporter in the periaqueductal gray matter. Neuroscience 2012, 223, 209–218. [Google Scholar] [CrossRef]
- Arnone, D.; Mumuni, A.N.; Jauhar, S.; Condon, B.; Cavanagh, J. Indirect evidence of selective glial involvement in glutamate-based mechanisms of mood regulation in depression: Meta-analysis of absolute prefrontal neuro-metabolic concentrations. Eur. Neuropsychopharmacol. 2015, 25, 1109–1117. [Google Scholar] [CrossRef]
- Henter, I.D.; de Sousa, R.T.; Zarate, C.A., Jr. Glutamatergic Modulators in Depression. Harv. Rev. Psychiatry 2018, 26, 307–319. [Google Scholar] [CrossRef]
- Seeburg, P.H. A-to-I editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Yuen, E.Y.; Liu, W.; Li, X.; Zhong, P.; Karatsoreos, I.N.; McEwen, B.S.; Yan, Z. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol. Psychiatry 2014, 19, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Novaes, L.S.; Dos Santos, N.B.; Perfetto, J.G.; Goosens, K.A.; Munhoz, C.D. Environmental enrichment prevents acute restraint stress-induced anxiety-related behavior but not changes in basolateral amygdala spine density. Psychoneuroendocrinology 2018, 98, 6–10. [Google Scholar] [CrossRef]
- Wang, M.; Ramasamy, V.S.; Samidurai, M.; Jo, J. Acute restraint stress reverses impaired LTP in the hippocampal CA1 region in mouse models of Alzheimer’s disease. Sci. Rep. 2019, 9, 10955. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xu, H.; Chang, M.; Lv, C.; Xue, W.; Song, Z.; Zhang, L.; Zhang, X.; Tian, X. Retigabine ameliorates acute stress-induced impairment of spatial memory retrieval through regulating USP2 signaling pathways in hippocampal CA1 area. Neuropharmacology 2018, 135, 151–162. [Google Scholar] [CrossRef]
- Bonini, D.; Mora, C.; Tornese, P.; Sala, N.; Filippini, A.; La Via, L.; Milanese, M.; Calza, S.; Bonanno, G.; Racagni, G.; et al. Acute Footshock Stress Induces Time-Dependent Modifications of AMPA/NMDA Protein Expression and AMPA Phosphorylation. Neural Plast. 2016, 2016, 7267865. [Google Scholar] [CrossRef] [Green Version]
- Caudal, D.; Rame, M.; Jay, T.M.; Godsil, B.P. Dynamic Regulation of AMPAR Phosphorylation In Vivo Following Acute Behavioral Stress. Cell. Mol. Neurobiol. 2016, 36, 1331–1342. [Google Scholar] [CrossRef]
- Jin, Y.; Kanno, T.; Nishizaki, T. Acute restraint stress impairs induction of long-term potentiation by activating GSK-3beta. Neurochem. Res. 2015, 40, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, G.; Jo, J.; Hogg, E.L.; Piers, T.; Kim, D.H.; Seaton, G.; Seok, H.; Bru-Mercier, G.; Son, G.H.; Regan, P.; et al. Acute stress causes rapid synaptic insertion of Ca2+ -permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain 2013, 136, 3753–3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, V.; Estil, J.B.; Chen, D.; Schrott, L.M.; Serrano, P.A. Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS ONE 2013, 8, e79077. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, Y.; Zhang, N.; Guo, Y.Y.; Feng, B.; Liu, S.B.; Zhao, M.G. Estrogen receptor GPR30 exerts anxiolytic effects by maintaining the balance between GABAergic and glutamatergic transmission in the basolateral amygdala of ovariectomized mice after stress. Psychoneuroendocrinology 2013, 38, 2218–2233. [Google Scholar] [CrossRef]
- Garcia-Keller, C.; Martinez, S.A.; Esparza, M.A.; Bollati, F.; Kalivas, P.W.; Cancela, L.M. Cross-sensitization between cocaine and acute restraint stress is associated with sensitized dopamine but not glutamate release in the nucleus accumbens. Eur. J. Neurosci. 2013, 37, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Caudal, D.; Godsil, B.P.; Mailliet, F.; Bergerot, D.; Jay, T.M. Acute stress induces contrasting changes in AMPA receptor subunit phosphorylation within the prefrontal cortex, amygdala and hippocampus. PLoS ONE 2010, 5, e15282. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Liu, W.; Karatsoreos, I.N.; Feng, J.; McEwen, B.S.; Yan, Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc. Natl. Acad. Sci. USA 2009, 106, 14075–14079. [Google Scholar] [CrossRef] [Green Version]
- Suenaga, T.; Morinobu, S.; Kawano, K.; Sawada, T.; Yamawaki, S. Influence of immobilization stress on the levels of CaMKII and phospho-CaMKII in the rat hippocampus. Int. J. Neuropsychopharmacol. 2004, 7, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Rosa, M.L.; Guimaraes, F.S.; Pearson, R.C.; Del Bel, E.A. Effects of single or repeated restraint stress on GluR1 and GluR2 flip and flop mRNA expression in the hippocampal formation. Brain Res. Bull. 2002, 59, 117–124. [Google Scholar] [CrossRef]
- Elhussiny, M.E.A.; Carini, G.; Mingardi, J.; Tornese, P.; Sala, N.; Bono, F.; Fiorentini, C.; La Via, L.; Popoli, M.; Musazzi, L.; et al. Modulation by chronic stress and ketamine of ionotropic AMPA/NMDA and metabotropic glutamate receptors in the rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110033. [Google Scholar] [CrossRef]
- Lin, M.; Hou, G.; Zhao, Y.; Yuan, T.F. Recovery of Chronic Stress-Triggered Changes of Hippocampal Glutamatergic Transmission. Neural Plast. 2018, 2018, 9360203. [Google Scholar] [CrossRef]
- Pillai, A.G.; Arp, M.; Velzing, E.; Lesuis, S.L.; Schmidt, M.V.; Holsboer, F.; Joels, M.; Krugers, H.J. Early life stress determines the effects of glucocorticoids and stress on hippocampal function: Electrophysiological and behavioral evidence respectively. Neuropharmacology 2018, 133, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.Y.; Hu, Z.L.; Zhang, D.Z.; Lu, W.; Zhou, J.; Wu, P.F.; Guan, X.L.; Han, Q.Q.; Deng, S.L.; Zhang, H.; et al. Rapid Antidepressant Effect of Hydrogen Sulfide: Evidence for Activation of mTORC1-TrkB-AMPA Receptor Pathways. Antioxid. Redox Signal. 2017, 27, 472–488. [Google Scholar] [CrossRef]
- Xia, B.; Chen, C.; Zhang, H.; Xue, W.; Tang, J.; Tao, W.; Wu, R.; Ren, L.; Wang, W.; Chen, G. Chronic stress prior to pregnancy potentiated long-lasting postpartum depressive-like behavior, regulated by Akt-mTOR signaling in the hippocampus. Sci. Rep. 2016, 6, 35042. [Google Scholar] [CrossRef] [Green Version]
- Gulemetova, R.; Drolet, G.; Kinkead, R. Neonatal stress augments the hypoxic chemoreflex of adult male rats by increasing AMPA receptor-mediated modulation. Exp. Physiol. 2013, 98, 1312–1324. [Google Scholar] [CrossRef]
- Esparza, M.A.; Bollati, F.; Garcia-Keller, C.; Virgolini, M.B.; Lopez, L.M.; Brusco, A.; Shen, H.W.; Kalivas, P.W.; Cancela, L.M. Stress-induced sensitization to cocaine: Actin cytoskeleton remodeling within mesocorticolimbic nuclei. Eur. J. Neurosci. 2012, 36, 3103–3117. [Google Scholar] [CrossRef] [Green Version]
- Ju, L.; Yang, J.; Zhu, T.; Liu, P.; Yang, J. BDNF-TrkB signaling-mediated upregulation of Narp is involved in the antidepressant-like effects of (2R,6R)-hydroxynorketamine in a chronic restraint stress mouse model. BMC Psychiatry 2022, 22, 182. [Google Scholar] [CrossRef]
- Lumsden, E.W.; Troppoli, T.A.; Myers, S.J.; Zanos, P.; Aracava, Y.; Kehr, J.; Lovett, J.; Kim, S.; Wang, F.H.; Schmidt, S.; et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc. Natl. Acad. Sci. USA 2019, 116, 5160–5169. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, K.; Fogaca, M.V.; Liu, R.J.; Duman, C.H.; Li, X.Y.; Chaki, S.; Duman, R.S. Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT1A receptor stimulation. Neuropsychopharmacology 2020, 45, 1725–1734. [Google Scholar] [CrossRef]
- Li, X.; Witkin, J.M.; Need, A.B.; Skolnick, P. Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell. Mol. Neurobiol. 2003, 23, 419–430. [Google Scholar] [CrossRef]

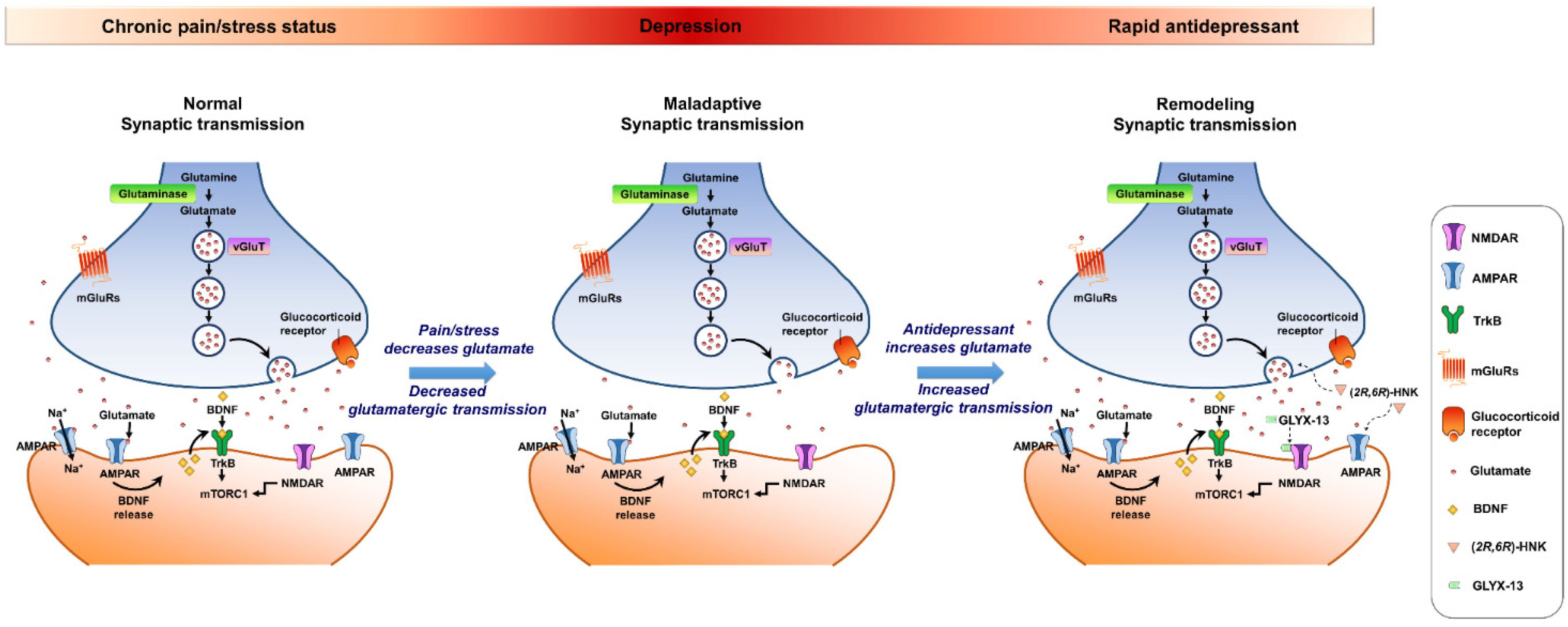
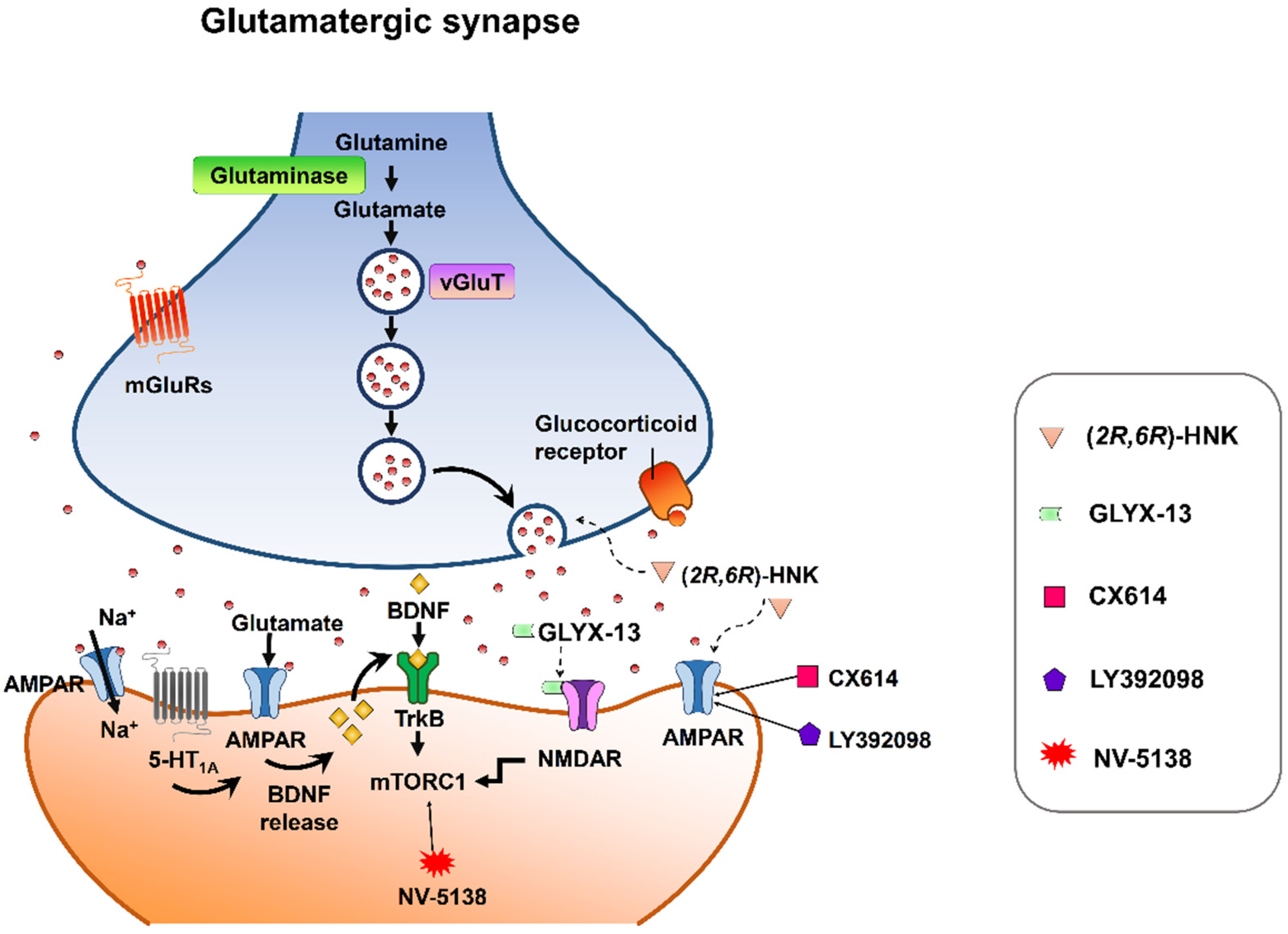

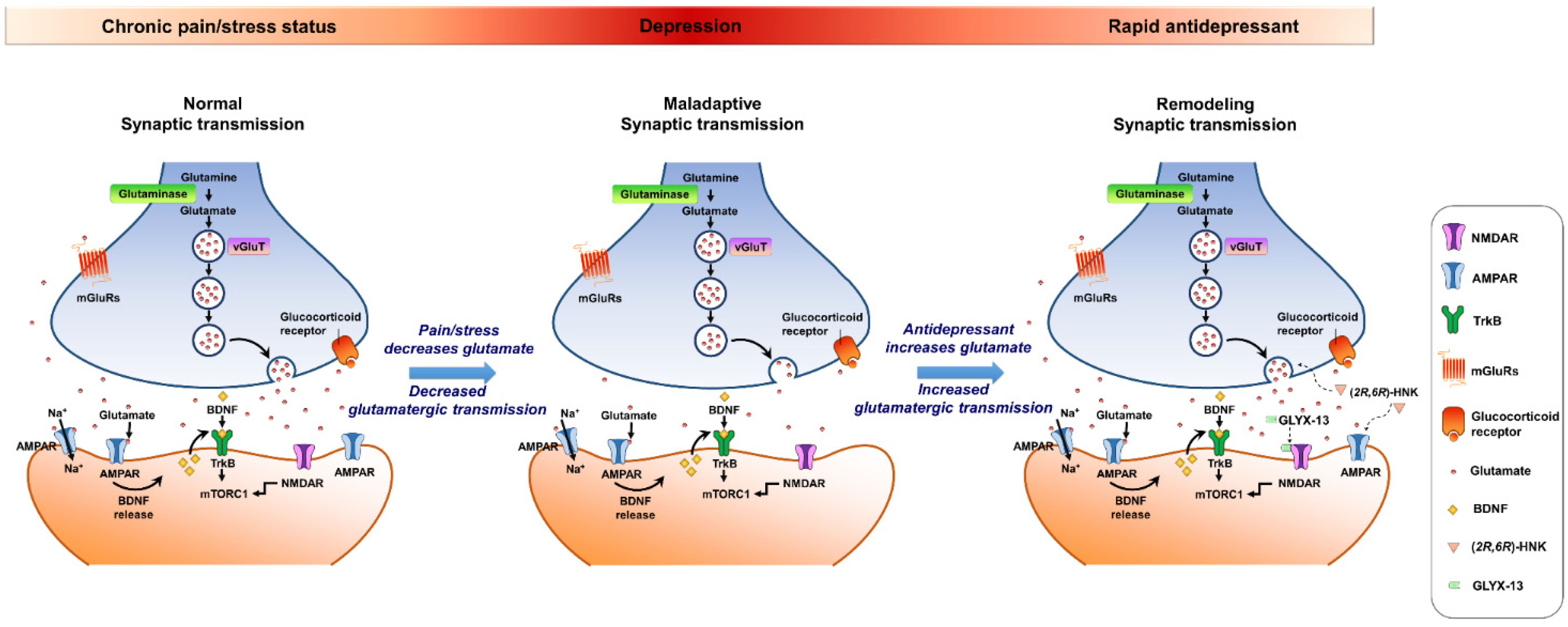
{kind=link}
{kind=link}
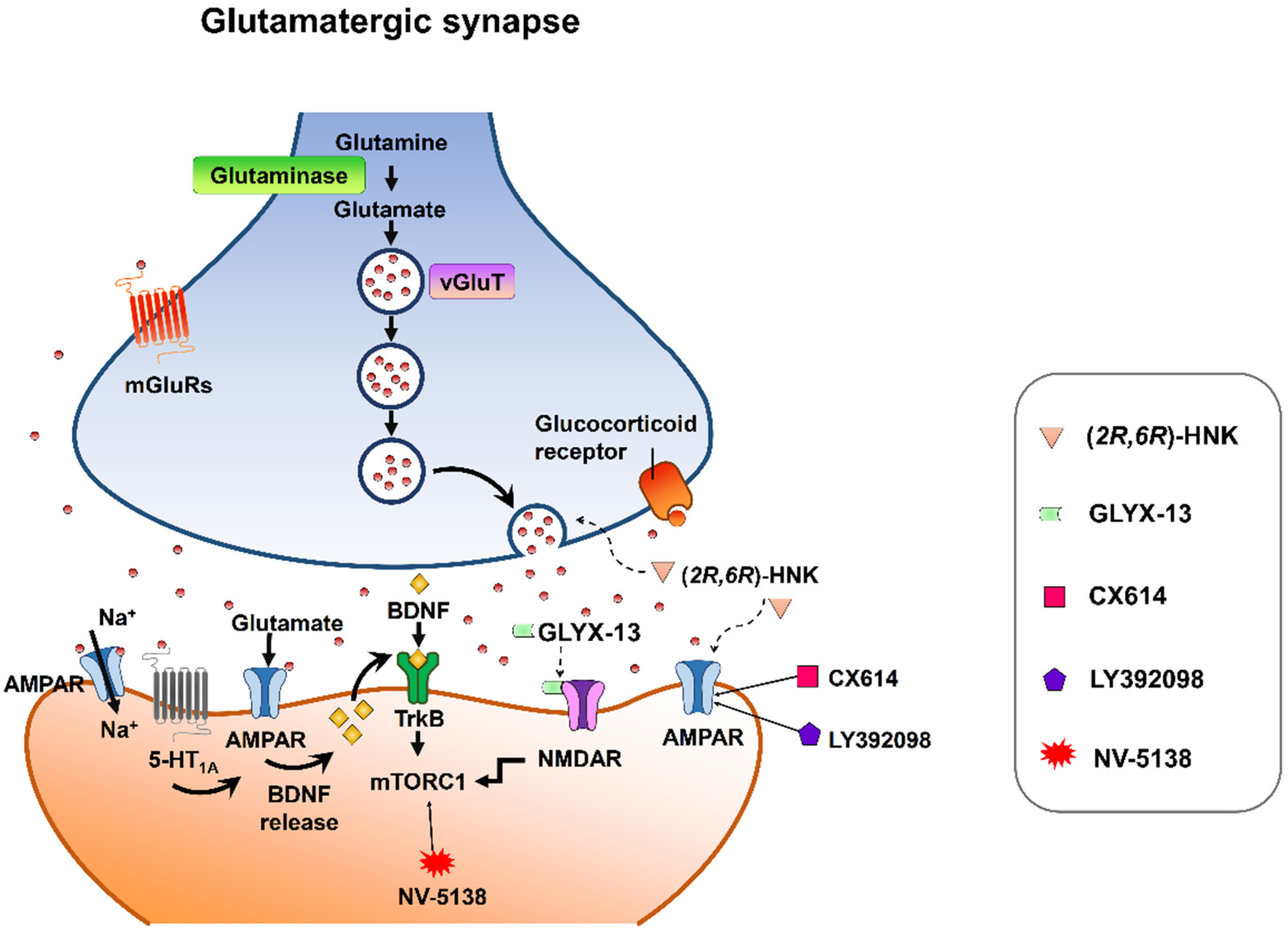{kind=link}
| Classification | Type of Stress | Brain Area | Effects on AMPA Receptor | Reference |
|---|---|---|---|---|
| Acute stress | Restraint stress for 2 h | Hippocampus | GluA1 subunit phosphorylation ↑ | [92] |
| Basolateral amygdala | GluA1 subunit phosphorylation - | [92] | ||
| Frontal cortex | GluA1 subunit phosphorylation - | [92] | ||
| Restraint stress for 30 min | Hippocampus | GluA1 subunit phosphorylation ↑ GluA1 expression ↑ | [93] | |
| Unsteady platform for acute stress | Hippocampus | GluA1 expression ↓ | [94] | |
| Acute footshock stress | Prefrontal and frontal cortex | GluA1 subunit phosphorylation ↑ GluA2 subunit phosphorylation ↑ | [95] | |
| Elevated platform stress | Amygdala | GluA1 subunit phosphorylation ↑ | [96] | |
| mPFC | GluA1 subunit phosphorylation ↑ | [96] | ||
| Hippocampus | GluA1 subunit phosphorylation ↑ | [96] | ||
| Acute restraint stress for 1 h | Hippocampus | GluA1 expression - GluA2 expression - | [97] | |
| Acute restraint stress for 30 min | Hippocampus | GluA1 subunit phosphorylation ↑ | [98] | |
| Elevated platform stress | Hippocampus | GluA2 expression ↓ | [99] | |
| Restraint or forced swimming | Amygdala | GluA1 subunit phosphorylation ↑GluA1 expression ↑ | [100] | |
| Acute restraint stress for 2 h | Nucleus accumbens | GluA1 expression ↑ | [101] | |
| Unsteady platform for 30 min | mPFC | Ser831-GluA1 phosphorylation ↓ Ser880-GluA2 phosphorylation ↑ | [102] | |
| Hippocampus | Ser831-GluA1 phosphorylation ↓ | [102] | ||
| Amygdala | Ser845-GluA1 phosphorylation ↑ Tyr876-GluA2 phosphorylation ↓ Ser880-GluA2 phosphorylation ↓ | [102] | ||
| Forced-swim stress | Prefrontal cortex | Surface GluA1 expression ↑ Surface GluA2 expression ↑ | [103] | |
| Immobilization stress for 45 min | Hippocampus | AMPA mRNA levels - | [104] | |
| Acute restraint stress for 6 h | Dentate gyrus | GluR2 flip mRNA expression↑ | [105] | |
| Chronic stress | Chronic mild stress | Hippocampus | AMPA mRNA - GluA2 expression ↑ | [106] |
| Chronic unpredictable mild stress | Hippocampus | GluA1 expression - GluA2 expression↑ GluA3 expression ↑ | [107] | |
| Early life Stress | Hippocampus | NMDA/AMPA ratio ↓ | [108] | |
| Chronic unpredictable mild stress | Hippocampus | GluA1 expression ↓ GluA2 expression ↓ GluA1 subunit phosphorylation ↓ | [109] | |
| Week chronic mild stress | Hippocampus | GluA1 expression ↓ | [110] | |
| Chronic unpredictable stress | Hippocampus | GluA1 expression ↓ | [44] | |
| Neonatal isolation stress | Paraventricular nucleus | AMPA binding sites ↑ | [111] | |
| Chronic immobilized stress | Nucleus accumbens | GluA1 expression ↑ | [112] | |
| Immobilization stress for 14 days | Hippocampus | AMPA mRNA levels - | [104] | |
| Chronic restraint stress for 21 days | Hippocampus CA1 | GluR1 flip mRNA expression ↓ | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.T.; Peng, W.-H.; Kan, H.-W.; Wu, C.-C.; Wang, D.-W.; Ho, Y.-C. Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor. Biomedicines 2022, 10, 1005. https://doi.org/10.3390/biomedicines10051005
Lee MT, Peng W-H, Kan H-W, Wu C-C, Wang D-W, Ho Y-C. Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor. Biomedicines. 2022; 10(5):1005. https://doi.org/10.3390/biomedicines10051005
Chicago/Turabian StyleLee, Ming Tatt, Wei-Hao Peng, Hung-Wei Kan, Cheng-Chun Wu, Deng-Wu Wang, and Yu-Cheng Ho. 2022. "Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor" Biomedicines 10, no. 5: 1005. https://doi.org/10.3390/biomedicines10051005
APA StyleLee, M. T., Peng, W.-H., Kan, H.-W., Wu, C.-C., Wang, D.-W., & Ho, Y.-C. (2022). Neurobiology of Depression: Chronic Stress Alters the Glutamatergic System in the Brain—Focusing on AMPA Receptor. Biomedicines, 10(5), 1005. https://doi.org/10.3390/biomedicines10051005