
Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature

 ,
,  , and
, and
Abstract
:1. Introduction
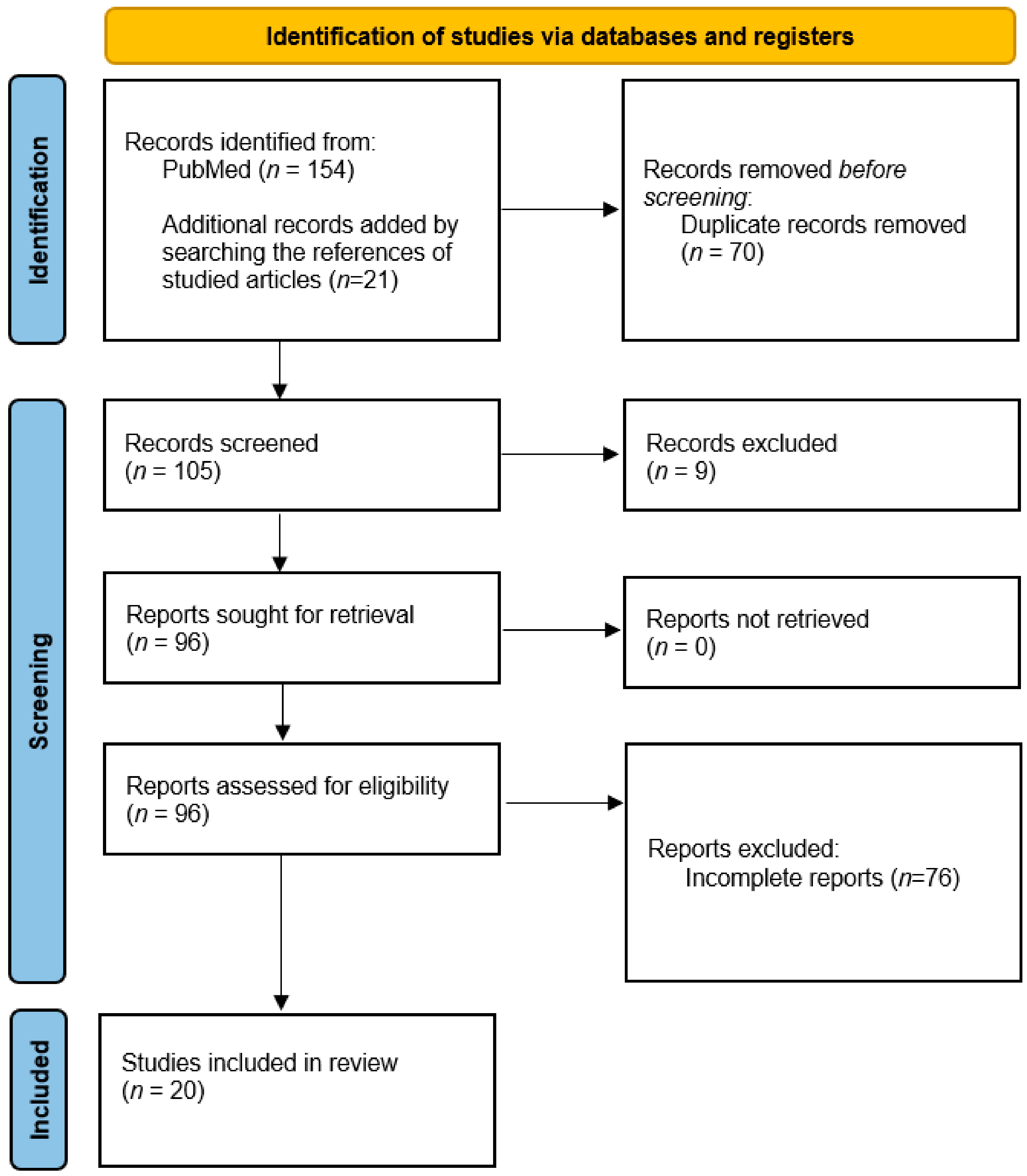
2. Materials and Methods
3. Results
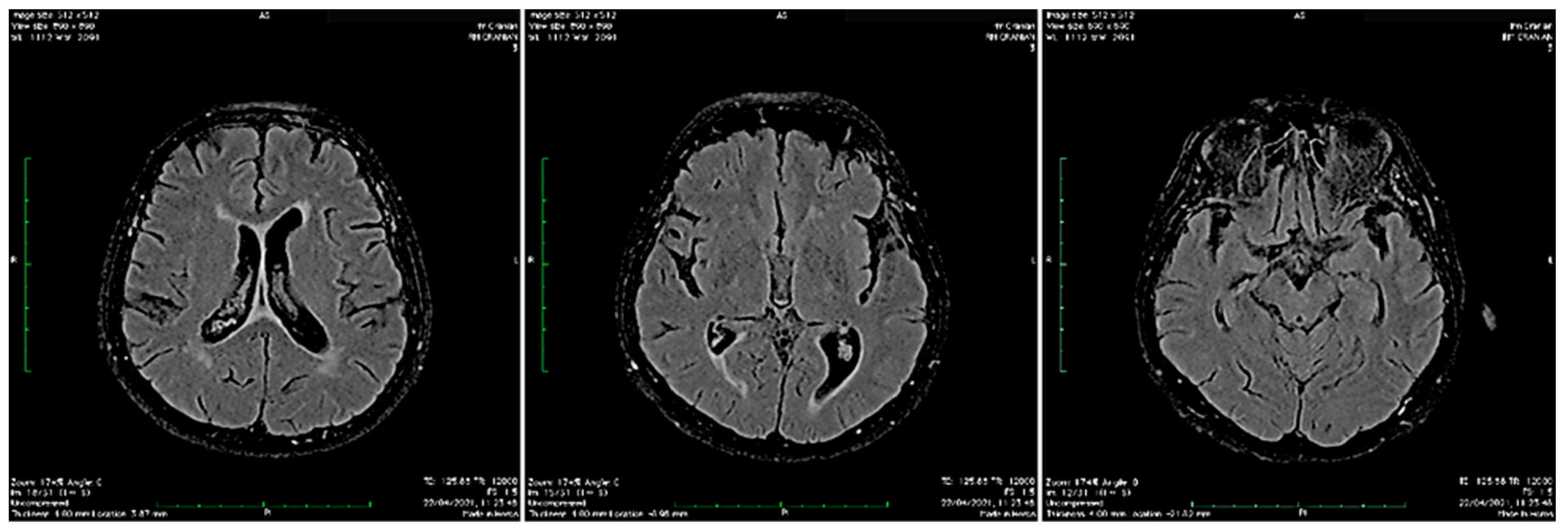
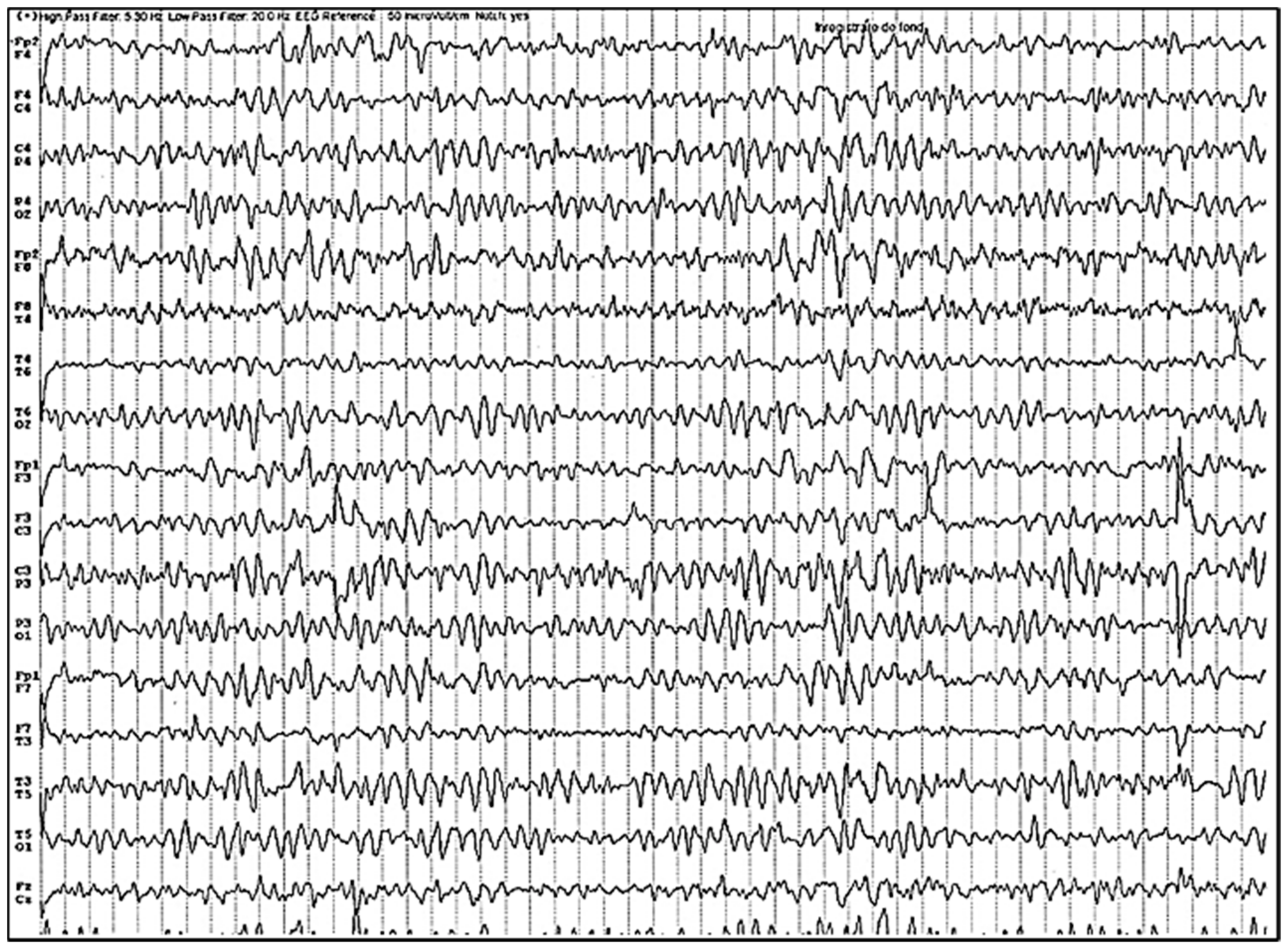
3.1. Our clinical experience
3.1.1. Literature Review
3.1.2. Long COVID and Neuro-COVID
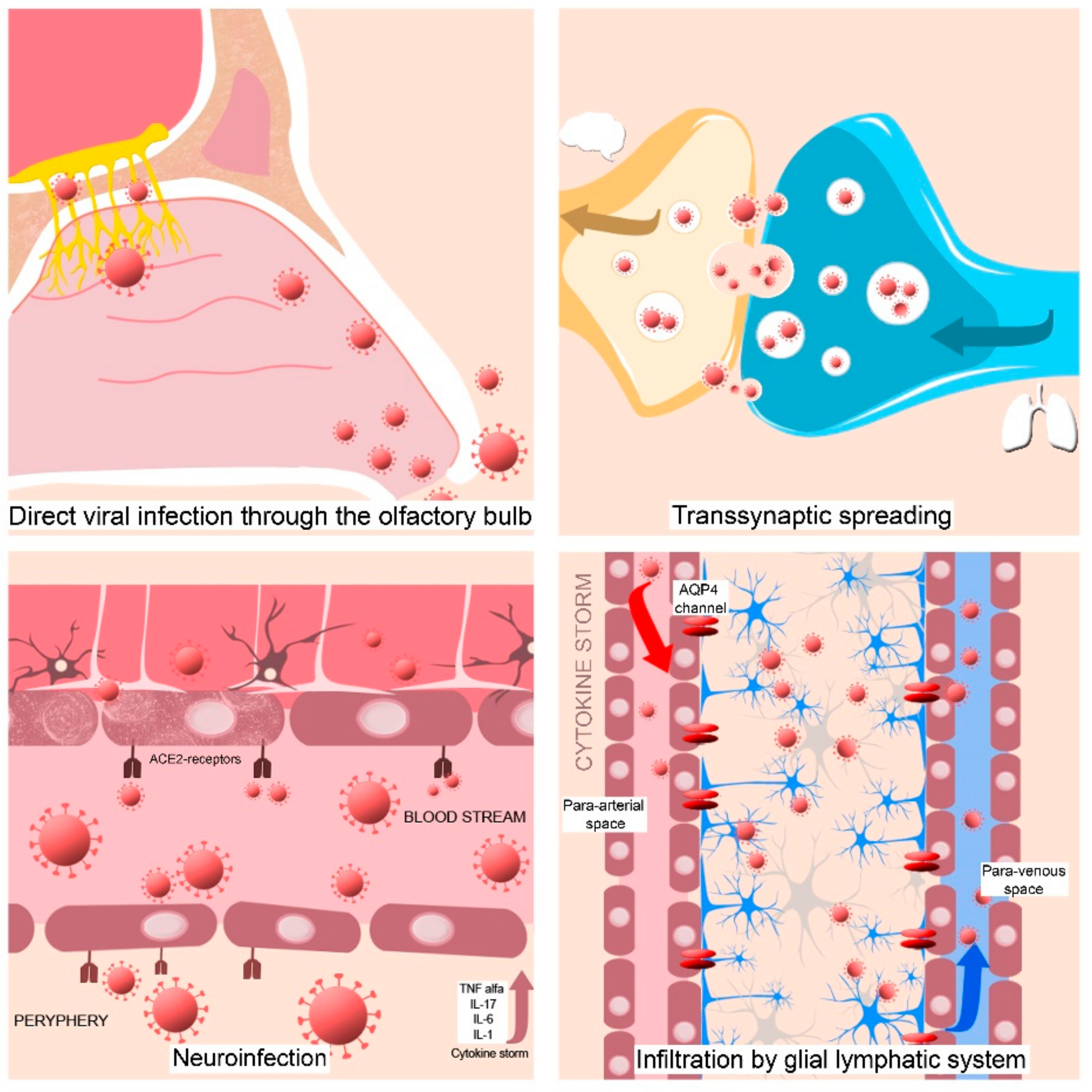
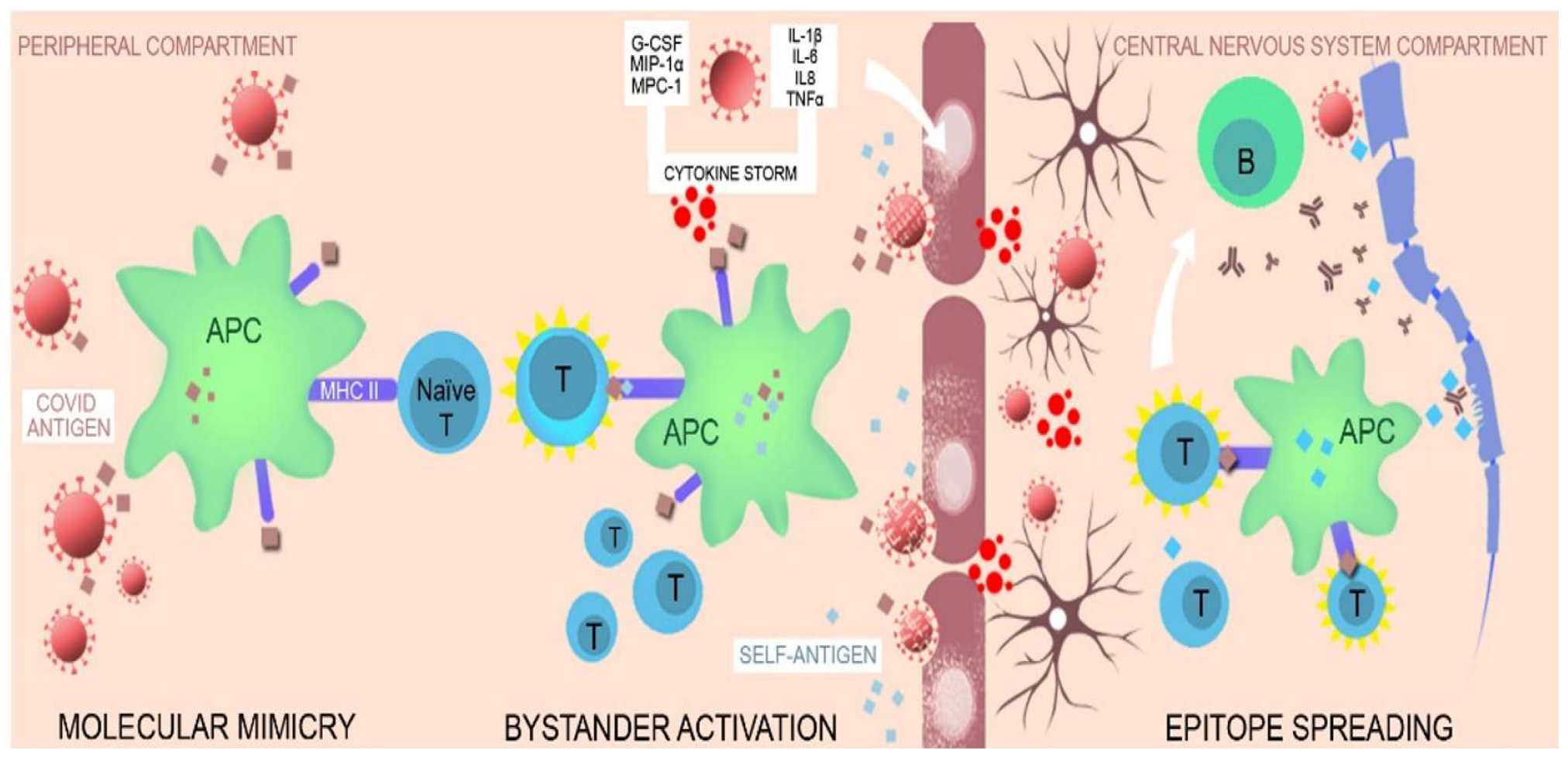
3.1.3. Pathophysiology of COVID-19 Neurologic Complications
- The virus could enter via the olfactory nerve and move, across the cribriform plate to the brainstem by retrograde transport and then disseminate in CNS tissue [8]. This theory can be sustained by persistent anosmia/and or dysgeusia that is present in some COVID-19 patients [58,69,70] suggesting a nasopharyngeal route for CNS infection [4]. The inflammation of the olfactory bulb and olfactory mucosa caused by SARS-CoV-2 infection can explain the anosmia and the presence of CSF in the subarachnoid space of the meninges adjacent to the olfactory bulb can perpetuate the viral spreading in the CNS [53].
- The virus could be distributed through systemic circulation and then in the cerebral blood flow. As a result of its interaction with the endothelial angiotensin-converting enzyme 2 (ACE2) receptors, it could penetrate the affected blood–brain barrier (BBB), which was compromised previously by the hyperactive immune responses [69].
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid-mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef] [PubMed]
- Lledó, G.M.; Sellares, J.; Brotons, C.; Sans, M.; Antón, J.D.; Blanco, J.; Bassat, Q.; Sarukhan, A.; Miró, J.M.; de Sanjosé, S.; et al. Post-acute COVID-19 syndrome: A new tsunami requiring a universal case definition. Clin. Microbiol. Infect. 2021, 28, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; Crabbio, M.; Magni, E.; De Giuli, V.; Caprioli, F.; Rifino, N.; Sessa, M.; Gennuso, M.; et al. SARS-CoV-2 related encephalopaties (ENCOVID) Study Group. Clinical Presentation and Outcomes of Severe Acute Respiratory Syndrome Coronavirus 2-Related Encephalitis: The ENCOVID Multicenter Study. J. Infect. Dis. 2021, 223, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Odolini, S.; Masciocchi, S.; Comelli, A.; Volonghi, I.; Gazzina, S.; Nocivelli, S.; Pezzini, A.; Focà, E.; Caruso, A.; et al. Steroid-Responsive Encephalitis in Coronavirus Disease 2019. Ann. Neurol. 2020, 88, 423–427. [Google Scholar] [CrossRef]
- Stoian, A.; Bălașa, R.; Grigorescu, B.L.; Maier, S.; Andone, S.; Cocuz, I.G.; Bajko, Z.; Filep, C.R.; Stoian, M. Guillain-Barré syndrome associated with Covid-19: A close relationship or just a coincidence? (Review). Exp. Ther. Med. 2021, 22, 916. [Google Scholar] [CrossRef]
- Stoian, A.; Bajko, Z.; Maier, S.; Cioflinc, R.A.; Grigorescu, B.L.; Moțățăianu, A.; Bărcuțean, L.; Balașa, R.; Stoian, M. High-dose intravenous immunoglobulins as a therapeutic option in critical illness polyneuropathy accompanying SARS-CoV-2 infection: A case-based review of the literature (Review). Exp. Ther. Med. 2021, 22, 1182. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host-Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, E.M.; Hundal, J.; Feterman, D.; Magaldi, J. Concomitant new diagnosis of systemic lupus erythematosus and COVID-19 with possible antiphospholipid syndrome. Just a coincidence? A case report and review of intertwining pathophysiology. Clin. Rheumatol. 2020, 39, 2811–2815. [Google Scholar] [CrossRef]
- Mosora, O.; Moroșanu, V.; Stoian, A.; Bălașa, R. Para-Infectious Acute Transverse Myelitis Following SARS-Cov2 Infection: A Case Report. Acta Marisiensis 2021, 67, 170–172. [Google Scholar] [CrossRef]
- Siow, I.; Lee, K.S.; Zhang, J.J.Y.; Saffari, S.E.; Ng, A. Encephalitis as a neurological complication of COVID-19: A systematic review and meta-analysis of incidence, outcomes, and predictors. Eur. J. Neurol. 2021, 28, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Zandifar, A.; Badrfam, R. COVID-19 and anti-N-methyl-d-aspartate receptor (anti-NMDAR) encephalitis: Are we facing an increase in the prevalence of autoimmune encephalitis? J. Med. Virol. 2021, 93, 1913–1914. [Google Scholar] [CrossRef] [PubMed]
- WHO New Policy Brief Calls on Decision-Makers to Support Patients as 1 in 10 Reports Symptoms of “Long COVID”. Available online: https://www.euro.who.int/en/health-topics/health-emergencies/coronavirus-covid-19/news/news/2021/2/new-policy-brief-calls-on-decision-makers-to-support-patients-as-1-in-10-report-symptoms-of-long-covid (accessed on 7 January 2022).
- Guilmot, A.; Slootjes, S.M.; Sellimi, A.; Bronchain, M.; Hanseeuw, B.; Belkhir, L.; Yombi, J.C.; De Greef, J.; Pothen, L.; Yildiz, H.; et al. Immune mediated neurological syndromes in SARS-CoV-2 infected patients. J. Neurol. 2021, 268, 751–757. [Google Scholar] [CrossRef]
- Grimaldi, S.; Lagarde, S.; Harlé, J.R.; Boucraut, J.; Guedj, E. Autoimmune Encephalitis Concomitant with SARS-CoV-2 Infection: Insight from 18 F-FDG PET Imaging and Neuronal Autoantibodies. J. Nucl. Med. 2020, 61, 1726–1729. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Garner, P. BMJ Opinion. Paul Garner: For 7 Weeks I Have Been through a Roller Coaster of Ill Health, Extreme Emotions, and Utter Exhaustion. 2020. Available online: https://blogs.bmj.com/bmj/2020/05/05/paul-garner-people-who-have-a-more-protracted-illness-need-help-to-understand-and-cope-with-the-constantly-shifting-bizarre-symptoms/ (accessed on 7 January 2022).
- Ayuso, L.L.; Rubio, P.T.; do Rosário, R.F.B.; Arroyo, M.L.G.; Sierra-Hidalgo, F. Bickerstaff encephalitis after COVID-19. J. Neurol. 2021, 268, 2035–2037. [Google Scholar] [CrossRef]
- Monti, G.; Giovannini, G.; Marudi, A.; Bedin, R.; Melegari, A.; Simone, A.M.; Santangelo, M.; Pignatti, A.; Bertellini, E.; Trenti, T.; et al. Anti-NMDA receptor encephalitis presenting as new onset refractory status epilepticus in COVID-19. Seizure 2020, 81, 18–20. [Google Scholar] [CrossRef]
- Panariello, A.; Bassetti, R.; Radice, A.; Rossotti, R.; Puoti, M.; Corradin, M.; Moreno, M.; Percudani, M. Anti-NMDA receptor encephalitis in a psychiatric Covid-19 patient: A case report. Brain Behav. Immun. 2020, 87, 179–181. [Google Scholar] [CrossRef]
- Bravo, G.A.; Torrentà, L.R.i. Anti-NMDA receptor encephalitis secondary to SARS-CoV-2 infection. Neurologia 2020, 35, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Manganotti, P.; Furlanis, G.; Ajčević, M.; Moras, C.; Bonzi, L.; Pesavento, V.; Stella, A.B. Intravenous immunoglobulin response in new-onset refractory status epilepticus (NORSE) COVID-19 adult patients. J. Neurol. 2021, 268, 3569–3573. [Google Scholar] [CrossRef] [PubMed]
- Sarigecili, E.; Arslan, I.; Ucar, H.K.; Celik, U. Pediatric anti-NMDA receptor encephalitis associated with COVID-19. Childs. Nerv. Syst. 2021, 37, 3919–3922. [Google Scholar] [CrossRef] [PubMed]
- Allahyari, F.; Hosseinzadeh, R.; Nejad, J.H.; Heiat, M.; Ranjbar, R. A case report of simultaneous autoimmune and COVID-19 encephalitis. J. Neurovirol. 2021, 27, 504–506. [Google Scholar] [CrossRef]
- Burr, T.; Barton, C.; Doll, E.; Lakhotia, A.; Sweeney, M. N-Methyl-d-Aspartate Receptor Encephalitis Associated With COVID-19 Infection in a Toddler. Pediatr. Neurol. 2021, 114, 75–76. [Google Scholar] [CrossRef]
- Peters, J.; Alhasan, S.; Vogels, C.B.F.; Grubaugh, N.D.; Farhadian, S.; Longbrake, E.E. MOG-associated encephalitis following SARS-COV-2 infection. Mult. Scler. Relat. Disord. 2021, 50, 102857. [Google Scholar] [CrossRef]
- Gaughan, M.; Connolly, S.; O’Riordan, S.; Tubridy, N.; McGuigan, C.; Kinsella, J.A. Pediatric Parainfectious Encephalitis Associated With COVID-19. Neurology 2021, 96, 541–544. [Google Scholar] [CrossRef]
- Oosthuizen, K.; Steyn, E.C.; Tucker, L.; Ncube, I.V.; Hardie, D.; Marais, S. SARS-CoV-2 Encephalitis Presenting as a Clinical Cerebellar Syndrome: A Case Report. Neurology 2021, 97, 27–29. [Google Scholar] [CrossRef]
- Vraka, K.; Ram, D.; West, S.; Chia, W.Y.E.; Kurup, P.; Subramanian, G.; Tan, H.J. Two Paediatric Patients with Encephalopathy and Concurrent COVID-19 Infection: Two Sides of the Same Coin? Case Rep. Neurol. Med. 2021, 2021, 6658000. [Google Scholar] [CrossRef]
- Ahsan, N.; Jafarpour, S.; Santoro, J.D. Myelin oligodendrocyte glycoprotein antibody encephalitis following severe acute respiratory syndrome coronavirus 2 in a pediatric patient. Clin. Exp. Pediatr. 2021, 64, 310–312. [Google Scholar] [CrossRef]
- Valadez-Calderon, J.; Navarro, A.O.; Rodriguez-Chavez, E.; Vera-Lastra, O. Co-expression of anti-NMDAR and anti-GAD65 antibodies. A case of autoimmune encephalitis in a post-COVID-19 patient. Neurologia 2021. [Google Scholar] [CrossRef] [PubMed]
- Durovic, E.; Bien, C.; Bien, C.G.; Isenmann, S. MOG antibody-associated encephalitis secondary to Covid-19: Case report. BMC Neurol. 2021, 21, 414. [Google Scholar] [CrossRef] [PubMed]
- McHattie, A.W.; Coebergh, J.; Khan, F.; Morgante, F. Palilalia as a prominent feature of anti-NMDA receptor encephalitis in a woman with COVID-19. J. Neurol. 2021, 268, 3995–3997. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Morales, A.E.; Urrutia-Osorio, M.; Camacho-Mendoza, E.; Rosales-Pedraza, G.; Dávila-Maldonado, L.; González-Duarte, A.; Herrera-Mora, P.; Ruiz-García, M. Neurological manifestations temporally associated with SARS-CoV-2 infection in pediatric patients in Mexico. Child’s Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2021, 37, 2305–2312. [Google Scholar] [CrossRef]
- McAlpine, L.S.; Lifland, B.; Check, J.R.; Angarita, G.A.; Ngo, T.T.; Pleasure, S.J.; Wilson, M.R.; Spudich, S.S.; Farhadian, S.F.; Bartley, C.M. Remission of Subacute Psychosis in a COVID-19 Patient with an Antineuronal Autoantibody After Treatment With Intravenous Immunoglobulin. Biol. Psychiatry 2021, 90, e23–e26. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Kim, S.S.; Lindsell, C.J.; Rose, E.B.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Erickson, H.L.; Steingrub, J.S.; Smithline, H.A.; et al. Symptom Duration and Risk Factors for Delayed Return to Usual Health Among Outpatients with COVID-19 in a Multistate Health Care Systems Network—United States, March–June 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 993–998. [Google Scholar] [CrossRef]
- Bertin, D.; Kaphan, E.; Weber, S.; Babacci, B.; Arcani, R.; Faucher, B.; Ménard, A.; Brodovitch, A.; Mege, J.L.; Bardin, N. Persistent IgG anticardiolipin autoantibodies are associated with post-COVID syndrome. Int. J. Infect. Dis. 2021, 113, 23–25. [Google Scholar] [CrossRef]
- Guedj, E.; Million, M.; Dudouet, P.; Tissot-Dupont, H.; Bregeon, F.; Cammilleri, S.; Raoult, D. F-FDG brain PET hypometabolism in post-SARS-CoV-2 infection: Substrate for persistent/delayed disorders? Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 592–595. [Google Scholar] [CrossRef]
- World Health Organization. A Clinical Case Definition of Post COVID-19 Condition by a Delphi Consensus, 6 October 2021; World Health Organization, 2021; Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-Post_COVID-19_condition-Clinical_case_definition-2021.1 (accessed on 4 January 2022).
- Günster, C.; Busse, R.; Spoden, M.; Rombey, T.; Schillinger, G.; Hoffmann, W.; Weber-Carstens, S.; Schuppert, A.; Karagiannidis, C. 6-month mortality and readmissions of hospitalized COVID-19 patients: A nationwide cohort study of 8679 patients in Germany. PLoS ONE 2021, 16, e0255427. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Heming, M.; Li, X.; Räuber, S.; Mausberg, A.K.; Börsch, A.L.; Hartlehnert, M.; Singhal, A.; Lu, I.N.; Fleischer, M.; Szepanowski, F.; et al. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal. Fluid Immun. 2021, 54, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396. [Google Scholar] [CrossRef] [PubMed]
- Afrin, L.B.; Weinstock, L.B.; Molderings, G.J. COVID-19 hyperinflammation and post-COVID-19 illness may be rooted in mast cell activation syndrome. Int. J. Infect. Dis. 2020, 100, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Mu, J.; Guo, J.; Lu, L.; Liu, D.; Luo, J.; Li, N.; Liu, J.; Yang, D.; Gao, H.; et al. New onset neurologic events in people with COVID-19 in 3 regions in China. Neurology 2020, 95, e1479–e1487. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef] [PubMed]
- Tansey, C.M.; Louie, M.; Loeb, M.; Gold, W.L.; Muller, M.P.; de Jager, J.; Cameron, J.I.; Tomlinson, G.; Mazzulli, T.; Walmsley, S.L.; et al. One-year outcomes and health care utilization in survivors of severe acute respiratory syndrome. Arch. Intern. Med. 2007, 167, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Wostyn, P. COVID-19 and chronic fatigue syndrome: Is the worst yet to come? Med. Hypotheses. 2021, 146, 110469. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Brison, E.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and Neurotropic Human Respiratory Coronaviruses: Potential Neurovirulent Agents in Humans. Adv. Exp. Med. Biol. 2014, 807, 75–96. [Google Scholar]
- Fisicaro, F.; Di Napoli, M.; Liberto, A.; Fanella, M.; Di Stasio, F.; Pennisi, M.; Bella, R.; Lanza, G.; Mansueto, G. Neurological Sequelae in Patients with COVID-19: A Histopathological Perspective. Int. J. Environ. Res. Public Health 2021, 18, 1415. [Google Scholar] [CrossRef]
- González-Pinto, T.; Luna-Rodríguez, A.; Moreno-Estébanez, A.; Agirre-Beitia, G.; Rodríguez-Antigüedad, A.; Ruiz-Lopez, M. Emergency Room Neurology in Times of COVID-19: Malignant Ischaemic Stroke and SARS-CoV-2 Infection. Eur. J. Neurol. 2020, 27, e35–e36. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; Troakes, C.; Hanley, B.; Osborn, M.; Richardson, M.P.; Hotopf, M.; Bullmore, E.; Everall, I.P. Invited Review: The Spectrum of Neuropathology in COVID-19. Neuropathol. Appl. Neurobiol. 2020, 47, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Reichard, R.R.; Kashani, K.B.; Boire, N.A.; Constantopoulos, E.; Guo, Y.; Lucchinetti, C.F. Neuropathology of COVID-19: A Spectrum of Vascular and Acute Disseminated Encephalomyelitis (ADEM)-like Pathology. Acta Neuropathol. 2020, 140, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.K.; Webster, R.K.; Smith, L.E.; Woodland, L.; Wessely, S.; Greenberg, N.; Rubin, G.J. The psychological impact of quarantine and how to reduce it: Rapid review of the evidence. Lancet 2020, 395, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Najjar, S.; Najjar, A.; Chong, D.J.; Pramanik, B.K.; Kirsch, C.; Kuzniecky, R.I.; Pacia, S.V.; Azhar, S. Central nervous system complications associated with SARS-CoV-2 infection: Integrative concepts of pathophysiology and case reports. J. Neuroinflamm. 2020, 17, 231. [Google Scholar] [CrossRef]
- Schett, G.; Sticherling, M.; Neurath, M.F. COVID-19: Risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 2020, 20, 271–272. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Madore, C.; Bordeleau, M.; Tian, L.; Verkhratsky, A. Neuropathobiology of COVID-19: The Role for Glia. Front. Cell. Neurosci. 2020, 14, 592214. [Google Scholar] [CrossRef] [PubMed]
- Cañas, C.A. The triggering of post-COVID-19 autoimmunity phenomena could be associated with both transient immunosuppression and an inappropriate form of immune reconstitution in susceptible individuals. Med. Hypotheses. 2020, 145, 110345. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.; Kozhaya, L.; Placek, L.; Gunter, C.; Yigit, M.; Hardy, R.; Plassmeyer, M.; Coatney, P.; Lillard, K.; Bukhari, Z.; et al. SARS-CoV-2 specific antibody and neutralization assays reveal the wide range of the humoral immune response to virus. Commun. Biol. 2021, 4, 129. [Google Scholar] [CrossRef]
- Churilov, L.P.; Kanduc, D.; Ryabkova, V.A. COVID-19: Adrenal response and molecular mimicry. Isr. Med. Assoc. J. 2021, 23, 618–619. [Google Scholar] [PubMed]
- Gasmi, A.; Tippairote, T.; Mujawdiya, P.K.; Benahmed, A.G.; Menzel, A.; Dadar, M.; Bjørklund, G. Neurological Involvements of SARS-CoV2 Infection. Mol. Neurobiol. 2021, 58, 944–949. [Google Scholar] [CrossRef]
- Giacomelli, A.; Pezzati, L.; Conti, F.; Bernacchia, D.; Siano, M.; Oreni, L.; Rusconi, S.; Gervasoni, C.; Ridolfo, A.L.; Rizzardini, G.; et al. Self-reported Olfactory and Taste Disorders in Patients with Severe Acute Respiratory Coronavirus 2 Infection: A Cross-sectional Study. Clin. Infect. Dis. 2020, 71, 889–890. [Google Scholar] [CrossRef] [Green Version]
- Steardo, L.; Steardo, L., Jr.; Zorec, R.; Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. 2020, 229, e13473. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020, 35, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Desforges, M.; Miletti, T.C.; Gagnon, M.; Talbot, P.J. Activation of human monocytes after infection by human coronavirus 229E. Virus Res. 2007, 130, 228–240. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, N.; Day, R.; Newcombe, J.; Talbot, P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000, 74, 8913–8921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhong, S.; Liu, J.; Li, L.; Li, Y.; Wu, X.; Li, Z.; Deng, P.; Zhang, J.; Zhong, N.; et al. Detection of severe acute respiratory syndrome coronavirus in the brain: Potential role of the chemokine mig in pathogenesis. Clin. Infect. Dis. 2005, 41, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.A.; Collins, A.; Cohen, M.E.; Duffner, P.K.; Faden, H. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 2004, 113, e73–e76. [Google Scholar]
- Chung, H.Y.; Neu, C.; Wickel, J.; Kuckertz, S.L.; Coldewey, S.M. Neurofilament Light Chain in Patients with COVID-19 and Bacterial Pneumonia. Ann. Neurol. 2021, 90, 174–175. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Palasca, O.; Santos, A.; Stolte, C.; Gorodkin, J.; Jensen, L.J. TISSUES 2.0: An integrative web resource on mammalian tissue expression. Database (Oxford) 2018, 2018, bay003. [Google Scholar] [CrossRef]
- Xia, H.; Lazartigues, E. Angiotensin-converting enzyme 2 in the brain: Properties and future directions. J. Neurochem. 2008, 107, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Milanetti, E.; Miotto, M.; Di Rienzo, L.; Nagaraj, M.; Monti, M.; Golbek, T.W.; Gosti, G.; Roeters, S.J.; Weidner, T.; Otzen, D.E.; et al. In-Silico Evidence for a Two Receptor Based Strategy of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 690655. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Furube, E.; Nakano, Y.; Morita, M.; Miyata, S. Microglia are continuously activated in the circumventricular organs of mouse brain. J. Neuroimmunol. 2019, 331, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar]
- Hughes, A.N.; Appel, B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 2020, 23, 1055–1066. [Google Scholar] [CrossRef]
- Najjar, S.; Pahlajani, S.; De Sanctis, V.; Stern, J.N.H.; Najjar, A.; Chong, D. Neurovascular Unit Dysfunction and Blood-Brain Barrier Hyperpermeability Contribute to Schizophrenia Neurobiology: A Theoretical Integration of Clinical and Experimental Evidence. Front. Psychiatry 2017, 8, 83. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflamm. 2013, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Wojkowska, D.W.; Szpakowski, P.; Glabinski, A. Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 1000. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef] [PubMed]
- Biesmans, S.; Meert, T.F.; Bouwknecht, J.A.; Acton, P.D.; Davoodi, N.; De Haes, P.; Kuijlaars, J.; Langlois, X.; Matthews, L.J.; Ver Donck, L.; et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat. Inflamm. 2013, 2013, 271359. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.L.; Herz, J.; Fernandes, A.; Rocha, J.; Sepodes, B.; Brito, M.A.; McGavern, D.B.; Brites, D. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J. Neuroinflamm. 2015, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Deczkowska, A. Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol. 2016, 37, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F.; Hsu, B.L.; Kimura, H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991, 28, 254–260. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Weaver, D.F. COVID-19 as a Trigger of Brain Autoimmunity. ACS Chem. Neurosci. 2021, 12, 2558–2561. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yong, V.W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 2019, 15, 704–717. [Google Scholar] [CrossRef]
- Tay, T.L.; Carrier, M.; Tremblay, M.È. Physiology of Microglia. Adv. Exp. Med. Biol. 2019, 1175, 129–148. [Google Scholar]
- Pascual, O.; Achour, S.B.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [Green Version]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lin, Y.C.; Wu, T.; Salgado, A.D.; Mexhitaj, I.; Wuest, S.C.; Romm, E.; Ohayon, J.; Goldbach-Mansky, R.; Vanderver, A.; et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J. Immunol. 2014, 192, 2551–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Schmidbauer, M.L.; Dimitriadis, K.; Otto, S.; Knier, B.; Niesen, W.D.; Hosp, J.A.; Günther, A.; Lindemann, S.; Nagy, G.; et al. Cerebrospinal fluid findings in COVID-19 patients with neurological symptoms. J. Neurol. Sci. 2020, 418, 117090. [Google Scholar] [CrossRef]
- Kremer, S.; Lersy, F.; de Sèze, J.; Ferré, J.C.; Maamar, A.; Carsin-Nicol, B.; Collange, O.; Bonneville, F.; Adam, G.; Martin-Blondel, G.; et al. Brain MRI Findings in Severe COVID-19: A Retrospective Observational Study. Radiology 2020, 297, E242–E251. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Slaats, J.; Oever, J.T.; van de Veerdonk, F.L.; Netea, M.G. IL-1β/IL-6/CRP and IL-18/ferritin: Distinct Inflammatory Programs in Infections. PLoS Pathog. 2016, 12, e1005973. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Neutrophil-to-lymphocyte ratio and lymphocyte-to-C-reactive protein ratio in patients with severe coronavirus disease 2019 (COVID-19): A meta-analysis. J. Med. Virol. 2020, 92, 1733–1734. [Google Scholar] [CrossRef] [Green Version]
- Moon, C. Fighting COVID-19 exhausts T cells. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440. [Google Scholar] [CrossRef]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, R.; Wilson, K.; Flanagan, K.L.; Jaworowski, A.; Plebanski, M. Adaptive Immunity and the Risk of Autoreactivity in COVID-19. Int. J. Mol. Sci. 2021, 22, 8965. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for t- and b-cell epitope prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemazee, D. Mechanisms of central tolerance for b cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Maeda, Y.; Nishikawa, H.; Sugiyama, D.; Ha, D.; Hamaguchi, M.; Saito, T.; Nishioka, M.; Wing, J.B.; Adeegbe, D.; Katayama, I.; et al. Detection of self-reactive cd8(+) t cells with an anergic phenotype in healthy individuals. Science 2014, 346, 1536–1540. [Google Scholar] [CrossRef]
- Richards, D.M.; Ruggiero, E.; Hofer, A.-C.; Sefrin, J.P.; Schmidt, M.; von Kalle, C.; Feuerer, M. The contained self-reactive peripheral t cell repertoire: Size, diversity, and cellular composition. J. Immunol. 2015, 195, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Meffre, E.; Wardemann, H. B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol. 2008, 20, 632–638. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, P.S.; DeFranco, A.L. Making and breaking tolerance. Curr. Opin. Immunol. 2002, 14, 744–759. [Google Scholar] [CrossRef]
- Jackson, S.R.; Yuan, J.; Berrien-Elliott, M.M.; Chen, C.L.; Meyer, J.M.; Donlin, M.J.; Teague, R.M. Inflammation programs self-reactive cd8+ t cells to acquire t-box-mediated effector function but does not prevent deletional tolerance. J. Leukoc. Biol. 2014, 96, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Beretta, S.; Piatti, M.; Karantzoulis, A.; Piatti, M.L.; Santoro, P.; Viganò, M.; Giovannelli, G.; Pirro, F.; Montisano, D.A.; et al. Guillain-Barré syndrome related to COVID-19 infection. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e741. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shen, D.; Zhou, H.; Liu, J.; Chen, S. Guillain-Barré syndrome associated with SARS-CoV-2 infection: Causality or coincidence? Lancet Neurol. 2020, 19, 383–384. [Google Scholar] [CrossRef]
- Zulfiqar, A.A.; Lorenzo-Villalba, N.; Hassler, P.; Andrès, E. Immune Thrombocytopenic Purpura in a Patient with Covid-19. N. Engl. J. Med. 2020, 382, e43. [Google Scholar] [CrossRef]
- Zanin, L.; Saraceno, G.; Panciani, P.P.; Renisi, G.; Signorini, L.; Migliorati, K.; Fontanella, M.M. SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir. 2020, 162, 1491–1494. [Google Scholar] [CrossRef]
- Palao, M.; Fernández-Díaz, E.; Gracia-Gil, J.; Romero-Sánchez, C.M.; Díaz-Maroto, I.; Segura, T. Multiple sclerosis following SARS-CoV-2 infection. Mult. Scler. Relat. Disord. 2020, 45, 102377. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef]
- Viner, R.M.; Whittaker, E. Kawasaki-like disease: Emerging complication during the COVID-19 pandemic. Lancet 2020, 395, 1741–1743. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients after Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Kichloo, A.; Aljadah, M.; Albosta, M.; Wani, F.; Singh, J.; Solanki, S. COVID-19 and Acute Lupus Pneumonitis: Diagnostic and Treatment Dilemma. J. Investig. Med. High Impact Case Rep. 2020, 8, 2324709620933438. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.B. Encephalitis lethargica and the influenza virus. II. The influenza pandemic of 1918/19 and encephalitis lethargica: Epidemiology and symptoms. J. Neural Transm. 2009, 116, 1295–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, Y.B.; Lim, Y.H.; Kim, K.J.; Park, K.S.; Park, Y.J. Respiratory viral infections and the risk of rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armangue, T.; Moris, G.; Cantarín-Extremera, V.; Conde, C.E.; Rostasy, K.; Erro, M.E.; Portilla-Cuenca, J.C.; Turón-Viñas, E.; Málaga, I.; Muñoz-Cabello, B.; et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 2015, 85, 1736–1743. [Google Scholar] [CrossRef] [Green Version]
- DiMaggio, D.; Anderson, A.; Bussel, J.B. Cytomegalovirus can make immune thrombocytopenic purpura refractory. Br. J. Haematol. 2009, 146, 104–112. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Chen, P.; Lin, Q.; Zhu, X.; Zhang, L.; Xue, X. Correlation between systemic lupus erythematosus and cytomegalovirus infection detected by different methods. Clin. Rheumatol. 2015, 34, 691–698. [Google Scholar] [CrossRef]
- Moon, U.Y.; Park, S.J.; Oh, S.T.; Kim, W.U.; Park, S.H.; Lee, S.H.; Cho, C.S.; Kim, H.Y.; Lee, W.K.; Lee, S.K. Patients with systemic lupus erythematosus have abnormally elevated epstein-barr virus load in blood. Arthritis Res. 2004, 6, R295–R302. [Google Scholar] [CrossRef] [Green Version]
- Yokochi, T.; Yanagawa, A.; Kimura, Y.; Mizushima, Y. High titer of antibody to the epstein-barr virus membrane antigen in sera from patients with rheumatoid arthritis and systemic lupus erythematosus. J. Rheumatol. 1989, 16, 1029–1032. [Google Scholar]
- Salmi, A.; Ziola, B.; Hovi, T.; Reunanen, M. Antibodies to coronaviruses oc43 and 229e in multiple sclerosis patients. Neurology 1982, 32, 292–295. [Google Scholar] [CrossRef]
- Stewart, J.N.; Mounir, S.; Talbot, P.J. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology 1992, 191, 502–505. [Google Scholar] [CrossRef]
- Talbot, P.J.; Paquette, J.S.; Ciurli, C.; Antel, J.P.; Ouellet, F. Myelin basic protein and human coronavirus 229e cross-reactive t cells in multiple sclerosis. Ann. Neurol. 1996, 39, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.M.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.K.; Wong, C.K.; Chan, P.K.S.; Ng, M.H.L.; Yu, L.M.; Hui, D.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Ng, M.H.; Li, C.K. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology 2005, 10, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abenza-Abildúa, M.J.; Ramírez-Prieto, M.T.; Moreno-Zabaleta, R.; Arenas-Valls, N.; Salvador-Maya, M.A.; Algarra-Lucas, C.; Rojo Moreno-Arrones, B.; Sánchez-Cordón, B.; de Luna, J.O.-R.; Jimeno-Montero, C.; et al. Neurological complications in critical patients with COVID-19. Neurología 2020, 35, 621–627. [Google Scholar] [CrossRef]
- Gammazza, A.M.; Légaré, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; de Macario, E.C.; Macario, A.J.; Cappello, F. Human molecular chaperones share with sars-cov-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef]
- Kanduc, D. From anti-sars-cov-2 immune responses to COVID-19 via molecular mimicry. Antibodies 2020, 9, 33. [Google Scholar] [CrossRef]
- Angileri, F.; Légaré, S.; Gammazza, A.M.; de Macario, E.C.; Macario, A.J.L.; Cappello, F. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with COVID-19? Br. J. Haematol. 2020, 190, e92–e93. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Teruel, M.; Alarcón-Riquelme, M.E. Genetics of systemic lupus erythematosus and Sjögren's syndrome: An update. Curr. Opin. Rheumatol. 2016, 28, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M. The autoimmune tautology. Arthritis Res. Ther. 2010, 12, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachoyiannopoulos, P.G.; Magira, E.; Alexopoulos, H.; Jahaj, E.; Theophilopoulou, K.; Kotanidou, A.; Tzioufas, A.G. Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann. Rheum. Dis. 2020, 79, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, M.; Wang, J.; Gao, J. Sars-Cov-2: Underestimated damage to nervous system. Travel Med. Infect. Dis. 2020, 36, 101642. [Google Scholar] [CrossRef]
- Dono, F.; Carrarini, C.; Russo, M.; De Angelis, M.V.; Anzellotti, F.; Onofrj, M.; Bonanni, L. New-onset refractory status epilepticus (NORSE) in post SARS-CoV-2 autoimmune encephalitis: A case report. Neurol. Sci. 2021, 42, 35–38. [Google Scholar] [CrossRef]
- Newsom-Davis, J.; Buckley, C.; Clover, L.; Hart, I.; Maddison, P.; Tüzüm, E.; Vincent, A. Autoimmune disorders of neuronal potassium channels. Ann. N. Y. Acad. Sci. 2003, 998, 202–210. [Google Scholar] [CrossRef]
- Montojo, M.T.; Petit-Pedrol, M.; Graus, F.; Dalmau, J. Clinical spectrum and diagnostic value of antibodies against the potassium channel related protein complex. Neurologia 2015, 30, 295–301. [Google Scholar] [CrossRef]
- Lai, M.; Huijbers, M.G.; Lancaster, E.; Graus, F.; Bataller, L.; Balice-Gordon, R.; Cowell, J.K.; Dalmau, J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: A case series. Lancet Neurol. 2010, 9, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, E.; Huijbers, M.G.; Bar, V.; Boronat, A.; Wong, A.; Martinez-Hernandez, E.; Wilson, C.; Jacobs, D.; Lai, M.; Walker, R.W.; et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann. Neurol. 2011, 69, 303–311. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, M.; Liu, L.; Chen, G.; Hou, Y.; Wang, M.; Li, J. Neuronal Surface Antibody Syndrome: A Review of the Characteristics of the Disease and Its Association with Autoantibodies. Neuroimmunomodulation 2020, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sagane, K.; Ishihama, Y.; Sugimoto, H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int. J. Biol. Sci. 2008, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.D.; Lee, S.; Jin, Z.; Wright, M.; Smith, S.E.; Anderson, M.P. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat. Med. 2009, 15, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Van Sonderen, A.; Petit-Pedrol, M.; Dalmau, J.; Titulaer, M.J. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat. Rev. Neurol. 2017, 13, 290–301. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blümcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Sonderen, A.; Thijs, R.D.; Coenders, E.C.; Jiskoot, L.C.; Sanchez, E.; de Bruijn, M.A.; van Coevorden-Hameete, M.H.; Wirtz, P.W.; Schreurs, M.W.; Smitt, P.A.S.; et al. Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. Neurology 2016, 87, 1449–1456. [Google Scholar] [CrossRef]
- Ariño, H.; Armangué, T.; Petit-Pedrol, M.; Sabater, L.; Martinez-Hernandez, E.; Hara, M.; Lancaster, E.; Saiz, A.; Dalmau, J.; Graus, F. Anti-LGI1-associated cognitive impairment: Presentation and long-term outcome. Neurology 2016, 87, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.W.; Lee, S.T.; Shin, J.W.; Moon, J.; Lim, J.A.; Byun, J.I.; Kim, T.J.; Lee, K.J.; Kim, Y.S.; Park, K.I.; et al. VGKC-complex/LGI1-antibody encephalitis: Clinical manifestations and response to immunotherapy. J. Neuroimmunol. 2013, 265, 75–81. [Google Scholar] [CrossRef]
- Gao, L.; Liu, A.; Zhan, S.; Wang, L.; Li, L.; Guan, L.; Zhao, X.; Zhang, X.; Wang, Y. Clinical characterization of autoimmune LGI1 antibody limbic encephalitis. Epilepsy Behav. 2016, 56, 165–169. [Google Scholar] [CrossRef]
- Finke, C.; Prüss, H.; Heine, J.; Reuter, S.; Kopp, U.A.; Wegner, F.; Bergh, F.T.; Koch, S.; Jansen, O.; Münte, T.; et al. Evaluation of Cognitive Deficits and Structural Hippocampal Damage in Encephalitis with Leucine-Rich, Glioma-Inactivated 1 Antibodies. JAMA Neurol. 2017, 74, 50–59. [Google Scholar] [CrossRef]
- Traka, M.; Goutebroze, L.; Denisenko, N.; Bessa, M.; Nifli, A.; Havaki, S.; Iwakura, Y.; Fukamauchi, F.; Watanabe, K.; Soliven, B.; et al. Girault. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J. Cell Biol. 2003, 162, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.; Gow, A. Tight junctions potentiate the insulative properties of small CNS myelinated axons. J. Cell Biol. 2008, 183, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Pinatel, D.; Hivert, B.; Boucraut, J.; Saint-Martin, M.; Rogemond, V.; Zoupi, L.; Karagogeos, D.; Honnorat, J.; Faivre-Sarrailh, C. Inhibitory axons are targeted in hippocampal cell culture by anti-Caspr2 autoantibodies associated with limbic encephalitis. Front. Cell. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körtvelyessy, P.; Bauer, J.; Stoppel, C.M.; Brück, W.; Gerth, I.; Vielhaber, S.; Wiedemann, F.R.; Heinze, H.J.; Bartels, C.; Bien, C.G. Complement-associated neuronal loss in a patient with CASPR2 antibody-associated encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, S.R.; Pettingill, P.; Kleopa, K.A.; Schiza, N.; Waters, P.; Mazia, C.; Zuliani, L.; Watanabe, O.; Lang, B.; Buckley, C.; et al. Morvan syndrome: Clinical and serological observations in 29 cases. Ann Neurol. 2012, 72, 241–255. [Google Scholar] [CrossRef]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010, 133, 2734–2748. [Google Scholar] [CrossRef]
- van Sonderen, A.; Ariño, H.; Petit-Pedrol, M.; Leypoldt, F.; Körtvélyessy, P.; Wandinger, K.P.; Lancaster, E.; Wirtz, P.W.; Schreurs, M.W.; Smitt, P.A.S.; et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016, 87, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Sunwoo, J.S.; Lee, S.T.; Byun, J.I.; Moon, J.; Shin, J.W.; Jeong, D.E.; Lee, G.H.; Jeong, S.H.; Shin, Y.W.; Jung, K.H.; et al. Clinical manifestations of patients with CASPR2 antibodies. J. Neuroimmunol. 2015, 281, 17–22. [Google Scholar] [CrossRef]
- Bien, C.G.; Mirzadjanova, Z.; Baumgartner, C.; Onugoren, M.D.; Grunwald, T.; Holtkamp, M.; Isenmann, S.; Kermer, P.; Melzer, N.; Naumann, M.; et al. Anti-contactin-associated protein-2 encephalitis: Relevance of antibody titres, presentation and outcome. Eur. J. Neurol. 2017, 24, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflamm. 2013, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Sánchez, C.M.; Díaz-Maroto, I.; Fernández-Díaz, E.; Sánchez-Larsen, Á.; Layos-Romero, A.; García-García, J.; González, E.; Redondo-Peñas, I.; Perona-Moratalla, A.B.; Del Valle-Pérez, J.A.; et al. Neurologic manifestations in hospitalized patients with COVID-19: The ALBACOVID registry. Neurology 2020, 95, e1060–e1070. [Google Scholar] [CrossRef] [PubMed]
- Stoian, A.; Moțățăianu, A.; Bărcuțean, L.; Maier, S.; Bajko, Z.; Voidăzan, S.; Fărcaș, A.; Bălașa, R. Understandig the mechanism of action of intravenous immunoglobulins: A ten years experience in treating Guillain Barré syndrome. Farmacia 2020, 68, 426–435. [Google Scholar] [CrossRef]
- Balasa, R.; Maier, S.; Barcutean, L.; Stoian, A.; Motataianu, A. The direct deleterious effect of Th17 cells in the nervous system compartment in multiple sclerosis and experimental autoimmune encephalomyelitis: One possible link between neuroinflammation and neurodegeneration. Rev. Rom. Med. Lab. 2020, 28, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Thibodeau, R.; Jafroodifar, A.; Quraeshi, S.; Lisi, M. SARS-CoV-2 infection leading to ischemic and hemorrhagic brain lesions and acute respiratory distress syndrome. Radiol. Case Rep. 2021, 16, 753–759. [Google Scholar] [CrossRef]
- Coolen, T.; Lolli, V.; Sadeghi, N.; Rovai, A.; Trotta, N.; Taccone, F.S.; Creteur, J.; Henrard, S.; Goffard, J.C.; Dewitte, O.; et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology 2020, 95, e2016–e2027. [Google Scholar] [CrossRef]
- Kihira, S.; Delman, B.N.; Belani, P.; Stein, L.; Aggarwal, A.; Rigney, B.; Schefflein, J.; Doshi, A.H.; Pawha, P.S. Imaging Features of Acute Encephalopathy in Patients with COVID-19: A Case Series. AJNR Am. J. Neuroradiol. 2020, 41, 1804–1808. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Age (Years), Sex | RT-PCR Test Swab | General and Respiratory Symptoms | Days between Infection and Neurological Onset | Neurological Symptoms | EEG | Brain CT/MRI | CSF | CSF RT-PCR | Autoantibodies | Immunomodulatory Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Guilmot et al., 2020 [16] | 80, M | P | Mild | NA | Seizures | Generalized slowing | Normal brain MRI | 9 cells/mm3 | N | CASPR2 positivity on CFS and blood | Corticosteroids, therapeutic plasma exchange | Seizure’s control |
| Guilmot et al., 2020 [16] | 71, NA | P | Mild | 5 after | Gait ataxia, akathisia, early delirium and choreiform involuntary movements of the upper limbs | Generalized slowing | Brain CT: normal | Normal | N | Serum anti-gangliosides antibodies (anti-GD1b IgG) (brainstem encephalitis) | NA | Good clinical evolution |
| Grimaldi et al., 2020 [17] | 72, M | P | Fever 38.5 °C; Thoracic scan: peripheral bilateral ground glass lesions, opacities | 17 after | Bilateral upper limb tremor, impossible walking, ataxia, dysarthria, upper limb dysmetria, diffuse myoclonus | Symmetric diffuse background slowing | Normal brain MRI | Normal cell count, proteins: 49 mg/dL, negative oligoclonal bands | N | Autoantibodies directed against the nuclei of Purkinje cells, striatal and hippocampal neurons evidenced by nerve tissue immunostaining | Corticosteroids, IVIg | Improvement of neurological condition, myoclonic seizures control |
| Ayuso et al., 2020 [20] | 72, F | P | Mild | 30 after | Slight inattention, disorientation, down beat nystagmus in all gaze positions, truncal ataxia, reflex myoclonus in the face and both arms | Normal | Brain MRI: hyperintense lesions in the caudal vermis and right flocculus with contrast enhancement in the floor of the fourth ventricle | 10 leukocyte/mm3, glucose: 70 mg/dL, absence of oligoclonal bands | NA | Anti GD1a antibodies (Bickerstaff encephalitis) | 1000 mg IV methylprednisolone daily (for 5 days) | A significant improvement |
| Monti et al., 2020 [21] | 50, M | P | Absent | 4 after | Confabulations and delirious ideas, then in evolution: focal motor seizures, impaired awareness, oro-facial dyskinesia, automatisms, refractory status epilepticus | General dezorganization, | Brain MRI: negative | 76 cells, oligoclonal bands; negative for fungi, viruses, bacteria | N | NMDAR antibody present in CSF | Metilprednisolone, IVIg, therapeutic plasma exchange | 4 months after onset: discharged in good condition, without neurological deficits |
| Panariello et al., 2020 [22] | 23, M | P | Fever, desaturation (90% O2 saturation in inhaled air), three weeks later with dysautonomia (fluctuations in respiratory rate, blood pressure, cardiac rhythm, body temperature) | Same time | Anxiety, psychomotor agitation, auditory hallucinations, persecutory delusions, global insomnia; three weeks later: non-verbal, non-responsive to commands; able to move the extremities and react to noxious stimuli | Theta activity (6 Hz), unstable, non-reactive to visual stimuli and without asymmetries | CT cerebral scan was negative for neuroanatomical acute anomalies | Hematic appearance with 960 (red and white cells)/mm3; glucose: 70 mg/dL, proteins: 65.4 mg/dL, Ab anti NMDAR: positive | N | Positive Ab anti NMDAR in CSF | High doses of dexamethasone and IVIg | Clinical condition in amelioration at the date of publishing the article |
| Alvarez-Bravo et al., 2020 [23] | 30, F | P | Fever at admission; later in evolution hypovolemic shock after a surgical intervention for teratoma; admitted to ICU | 3 before | Paranoid ideation, psychomotor agitation, visual hallucinations, dysarthria; three days later: focal and generalized seizures; in evolution: decreased level of consciousness, generalized choreo-dystonic movements, blepharoclonus | Epileptic discharges in the left frontotemporal region; in evolution: delta brush pattern | A brain CT scan at admission was normal; brain MRI study performed a few days later: hyperintensities in the left hippocampus. | WBC: 44/mm3 (90% lymphocytes); proteins: 54.5 mg/dL | N | Anti NMDAR antibody positive in serum and CSF | Methylprednisolone, IVIg; later due to lack of appropriate response: rituximab | Discharged 70 days later with cognitive sequelae, memory disorders, emotional lability and sent to rehabilitation |
| Manganotti et al., 2021 [24] | 37, M | P | Severe, ICU admission with respiratory support | Same time | Convulsive status epilepticus | Persistent generalized epileptic discharges | Brain CT scan: normal; Brain MRI scan: normal, negative contrast enhancement | 1 mononuclear WBC, protein: 56.7 mg/dL, glucose: 66.1 mg/dL | N | Anti amphiphysin antibody | IVIg 0.4 mg/kg/5 days | Complete clinical recovery, free of seizures and EEG normalization, no respiratory support |
| Sarigecili et al., 2021 [25] | 7, M | P | Absent | 3 before | Behavioral and mood changes, encephalopathy, abnormal movements, seizures, autonomic instability, ataxia | Widespread delta waves | Brain MRI: normal | Normal except NMDAR antibodies | N | NMDAR antibody in CSF | IVIg, TPE, steroids (in this order) | Significant improvement to an ambulatory state |
| Allahyari et al., 2021 [26] | 18, F | P | Shortness of breath, dry coughs | Same time | Mood changes, depression, anhedonia, lack of concentration, seizures | NA | Brain CT at the beginning: brain edema; brain MRI after 3 days: normal | 13.900 WBC/mm3 (87% neutrophils) | P | NMDAR antibody in CSF | Corticosteroids, IVIg | Discharged with full recovery |
| Burr et al., 2021 [27] | 2, F | P | Absent respiratory symptoms; general symptoms: fever, reduced oral intake, constipation | Same time | Poor sleep, fussiness, no talking, hyperkinetic movements of the arms, legs, head, seizures | NA | Brain MRI (native and with contrast): normal | 7 leucocytes/mL (89% lymphocytes); glucose: 56 mg/dL; proteins: 25 mg/dL | N | NMDAR antibody positivity in the serum and CSF | 5 days: intravenous methylprednisolone (30 mg/kg/day) followed by IVIg 2 mg/kg | Gradually resolution of the abnormal movements and encephalopathy; return to base line 2 weeks after discharge |
| Peters et al., 2021 [28] | 23, M | P | Absent | 14 after | Left sided headache and dysesthesias; after 5 weeks: personality changes, cognitive slowing, mild inattention, delayed recall, decreased verbal fluency | Epileptiform spikes and left posterior temporal rhythmic delta activity, | Brain MRI: normal; brain MRI repeated after 2 weeks: left hemispheric leptomeningeal enhancement; diffuse left hemispheric hyperintensity on FLAIR | 1 WBC; proteins: 36 mg/dL; glucose: 78 mg/dL; Repeated LP after 2 weeks: 57 WBS/mm3 (50% lymphocytes) | N | Serum positivity for MOG IgG antibody and CSF negativity | Methylprednisolone 1 gr/day (5 days) and then oral steroid taper | Improvement of the cognitive symptoms, resolution of the headache; total resolution of the symptoms in 8 weeks. |
| Gaughan et al., 2021 [29] | 16, F | P | Fever, sore throat, tachycardia | 3 after | At admission: insomnia, anorexia, visual and auditory hallucinations, ritualistic behaviors, paranoia; mutism five days later with no voluntary activity; fecal and urinary incontinence; bilateral limb rigidity and tremor in evolution | An excess of delta and theta activity, more expressed in the right temporal derivations | Brain MRI: two tiny T2/FLAIR hyperintensities located in centrum semiovale bilaterally, without diffusion restriction and without contrast enhancement | WBC: 2 cells/ mm3; proteins: 43 mg/dL; glucose: 2.9 mmol/L | N | Anti-GAD antibody transiently positive in serum, negative in CSF | IVIg 0.4 mg/kg/day (5 days), followed by methylprednisolone 1 g per day, 3 consecutive days followed by a second course of IVIg | Discharged at home on day 98 after admission with significant cognitive and physical difficulties and sent to rehabilitation |
| Oosthuizen et al., 2021 [30] | 52, M | D0: N D17: P | Tachypnea (20 bpm), fever (37.7 °C) | 17 before | Progressive gait instability, multidirectional gaze-evoked nystagmus, truncal ataxia, dysatria | EEG at admission: normal | Brain CT at admission: central midbrain hypodensity Brain MRI: characteristic for brainstem encephalitis | CSF: pleocytosis (49 lymphocytes/ mm3, 2 PMN/ mm3); proteins: 37 mg/dL, glucose: 3.6 mmol/L | P | Onconeural antibodies positive for amphiphysin in serum | Prednisone (1 mg/kg/day) | Discharged on day 36, able to walk independently with a mild emotional lability |
| Vraka et al., 2021 [31] | 1, F | P | Fever, hypertension | 3 before | Altered consciousness, seizures, drowsy, hypotonic, swallow difficulties, in evolution decorticate posturing and GCS 5 points | EEG: diffuse slow wave background activity, no epileptiform discharges | Brain CT: biemispheric white matter hypodensities; Brain MRI: bilateral widespread white matter hypersignal abnormalities (including splenium corpus callosum, thalamus and pons) | WBC: 10/mm3 | N | MOG antibody positive in serum | Steroid therapy | At discharge the patient was able to sit and walk a few steps, eat and drink normally but with cortical visual impairment with gradual improvement after four months |
| Ahsan et al., 2021 [32] | 7, F | D0:N D9: P (Ab IgG) | Abdominal pain, fever | Same time | Status epilepticus, aphasia, encephalopathy, prolonged Todd’s paralysis; after o week: headache, dysarthria, altered mental status | EEG: cerebral slowing with left focal slowing | Brain MRI: cortical edema, peri Rolandic and posterior parietal lobe restricted diffusion | CSF at admission, WBC: 132/mm3 (64% lymphocytes), proteins: 54 mg/dL, glucose 73 mg/dL | N | MOG antibody positive in serum 1:40 at admission and 1:100 after 7 days | IVIg 2 g/kg over 3 days | She was discharged with improved condition, free of seizures with mild dysarthria |
| Valadez-Calderon et al., 2021 [33] | 28, M | P | Mild symptoms of COVID-19 | 14 after | Somnolence, incoherent speech, auditory hallucinations, suicidal ideation, generalized tonic-clonic seizures, catatonic symptoms; two days later: status epilepticus | Subcortical dysfunction in frontal, temporal and occipital regions | Brain MRI: hyperintensities in the bilateral anterior cingulate cortex and temporal lobes | NA | N | NMDAR and GAD65/67 antibody positive in serum and CSF | Methylprednisolone 1 g/daily (5 days), followed by IVIg 0.4 g/kg/day (5 days) | Clinical improvement; at six weeks follow-up: still presents mood changes, irritability, agitation episodes |
| Durovic et al., 2021 [34] | 22, M | P | Fever, general weakness | 3 after | Severe headache, neck stiffness, a loss of smell and taste; days 16: mild impairment in executive functions | NA | Brain MRI: multiple disseminated T2 and FLAIR hyperintensities, no contrast enhancement | Cells: 31/mm3; proteins: 39.9 mg/dL, glucose: 64 mg/dL, lactate 11.9 mg/L | N | MOG (1:640) and mGluR1 (1:40) antibody positive in serum and CSF | Methylprednisolone 1 g/daily (5 days) | Two months later the patient presents no residual symptoms |
| McHattie et al., 2021 [35] | 53, F | D0: N D14: P | Fever, myalgia; later in evolution: hypoxemia requiring oxygen therapy and transfer in ICU; day 17: dysautonomia (hypotension, bradycardia) | 14 before | Palilalia at admission, after three days: confusion, urinary retention then severe echolalia, echopraxia, behavioral disinhibition; focal seizures, left side weakness | Slow activity, no epileptiform discharges | Brain CT with intravenous contrast at admission: normal Brain MRI: hyperintensity in the left amygdala, left anterior putamen, subtle changes in the left amygdala (FLAIR) | WBC: 141/mm3 (100% lymphocytes); | N | NMDAR antibody positive in CSF (1:100), negative in serum | IV and oral steroids, IVIg, Tocilizumab | One month later: remission of palilalia and seizures, improvement of cognitive functions, left side weakness |
| Sánchez-Morales et al., 2021 [36] | 14, M | N | None | NA | Altered behavior and mental status, orolingual dyskinesias, insomnia, seizures | NA | Performed, result NA | Cells: 2/mm3, proteins 23 mg/dL | P | NMDAR antibody positive in CSF | Methylprednisolone, IVIg | A partial recovery of the neurologic symptoms with seizures control but with psychiatric symptoms persistence |
| McAlpine et al., 2021 [37] | 30, M | P | Fever and malaise | Gradually, after a few days | Initially hypersomnia, then insomnia, hallucinations, anxiety; in evolution: cognitive slowing, flat affect, akathisia | 12 h video EEG: normal | Brain CT: normal; Brain MRI (native and with gadolinium): unremarkable | Normal | N | Antineural autoantibodies evidenced in CSF by immunostaining | IVIg (2 g/kg, over 3 days) | Good evolution, regression of the symptoms |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoian, A.; Stoian, M.; Bajko, Z.; Maier, S.; Andone, S.; Cioflinc, R.A.; Motataianu, A.; Barcutean, L.; Balasa, R. Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature. Biomedicines 2022, 10, 774. https://doi.org/10.3390/biomedicines10040774
Stoian A, Stoian M, Bajko Z, Maier S, Andone S, Cioflinc RA, Motataianu A, Barcutean L, Balasa R. Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature. Biomedicines. 2022; 10(4):774. https://doi.org/10.3390/biomedicines10040774
Chicago/Turabian StyleStoian, Adina, Mircea Stoian, Zoltan Bajko, Smaranda Maier, Sebastian Andone, Roxana Adriana Cioflinc, Anca Motataianu, Laura Barcutean, and Rodica Balasa. 2022. "Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature" Biomedicines 10, no. 4: 774. https://doi.org/10.3390/biomedicines10040774
APA StyleStoian, A., Stoian, M., Bajko, Z., Maier, S., Andone, S., Cioflinc, R. A., Motataianu, A., Barcutean, L., & Balasa, R. (2022). Autoimmune Encephalitis in COVID-19 Infection: Our Experience and Systematic Review of the Literature. Biomedicines, 10(4), 774. https://doi.org/10.3390/biomedicines10040774