
Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultivation
2.2. Melanoma EVs Isolation
2.3. Melanoma EVs Characterization
2.4. Real-Time PCR for mRNA Detection
2.5. Real-Time PCR for miRNA Detection
2.6. Western Blotting
2.7. Influence of EVs on Cell Proliferation
2.8. Wound Healing (Scratch) and Invasion Assays
2.9. Flow Cytometry
2.10. Analysis of Intracellular Pathways Activity Using Bio-Plex Magnetic Assay
2.11. EGFR Knockdown
2.12. Analysis of Cytokine Release by Keratinocytes
2.13. TCGA Database Analysis
2.14. Statistical Analysis
3. Results
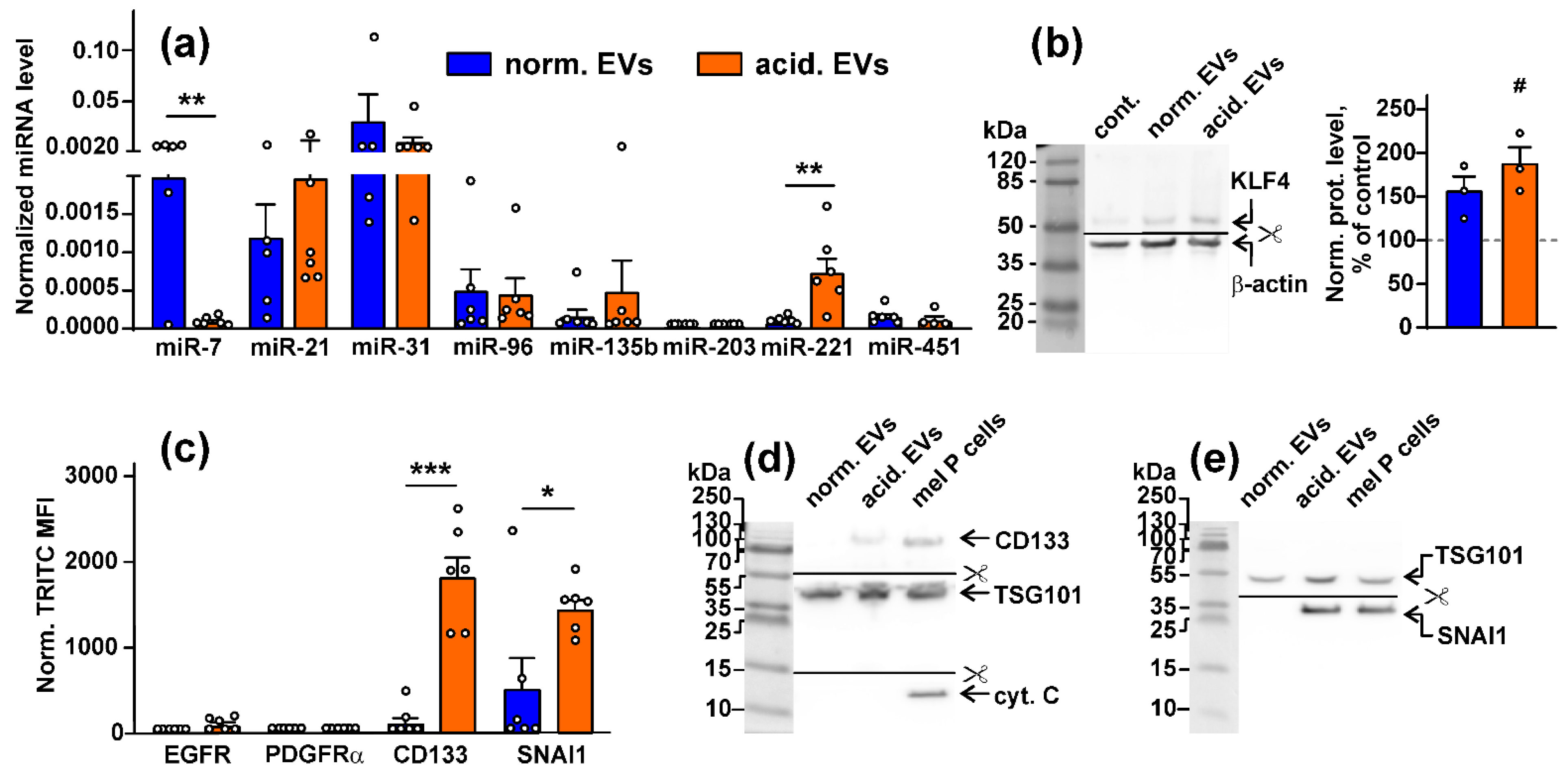
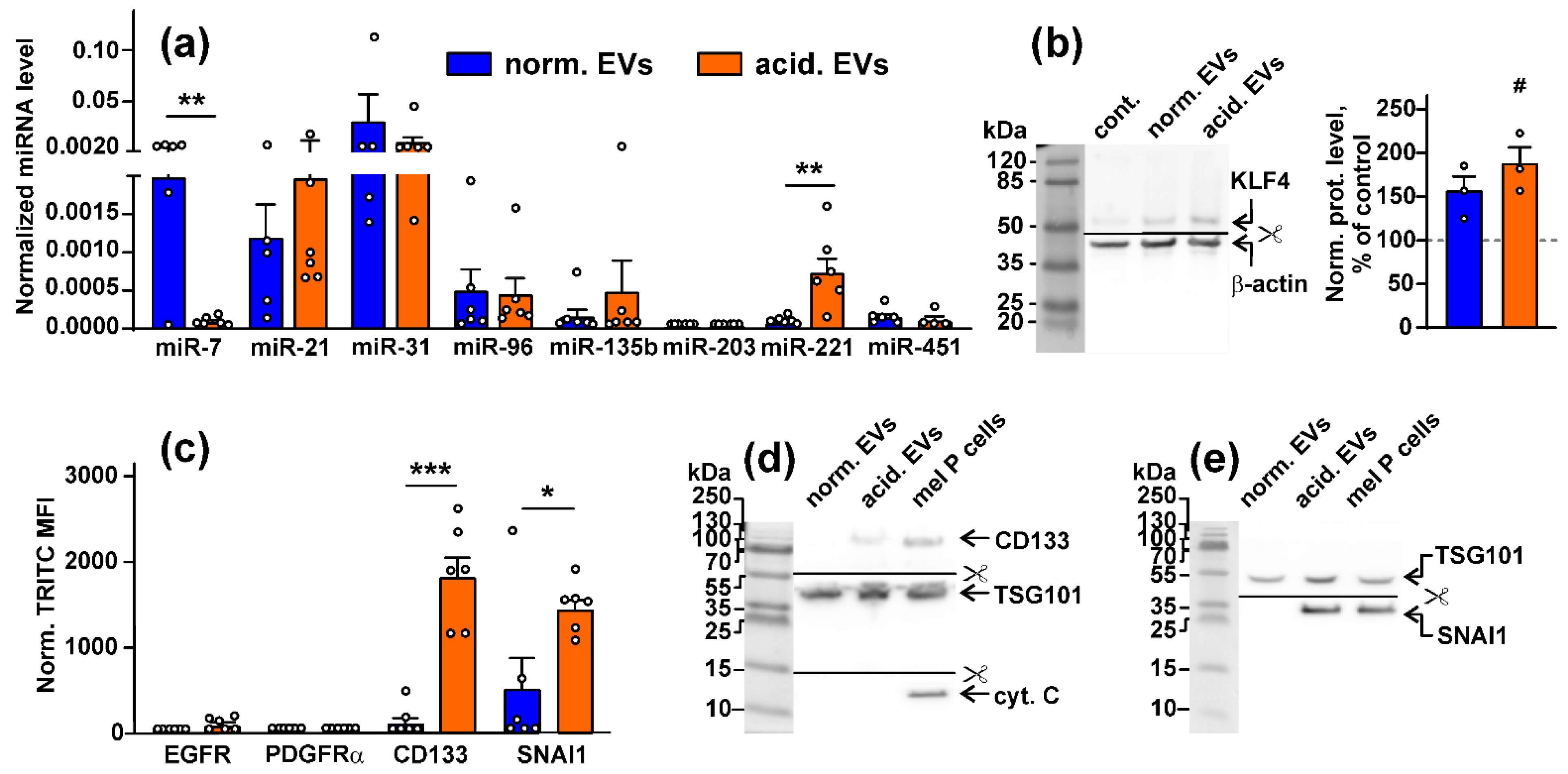
3.1. Acidification of Metastatic Melanoma Environment Makes EVs More Prooncogenic
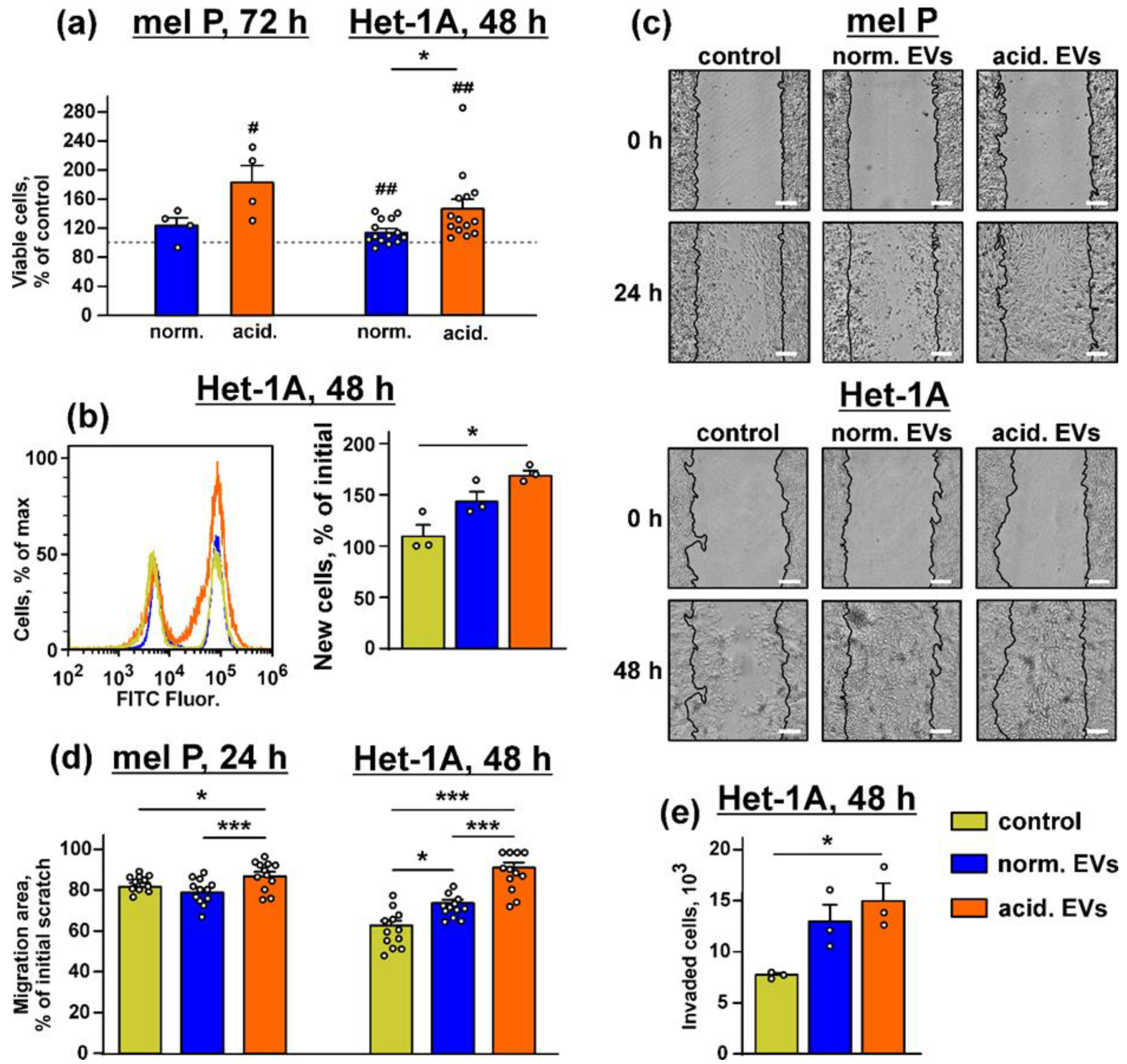
3.2. Metastatic Melanoma EVs Stimulate Growth and Migration of Metastatic Melanoma Cells and Normal Keratinocytes
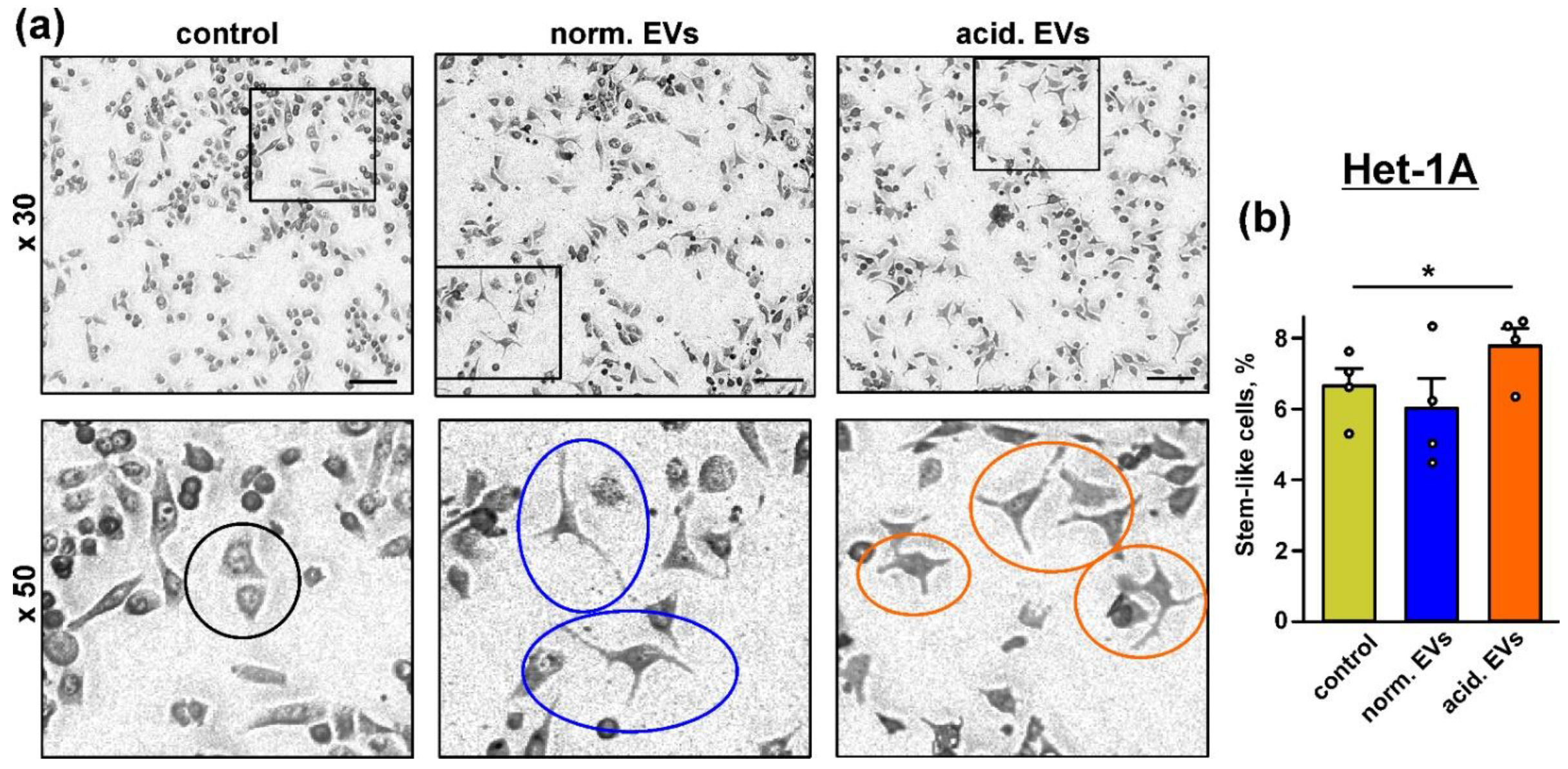
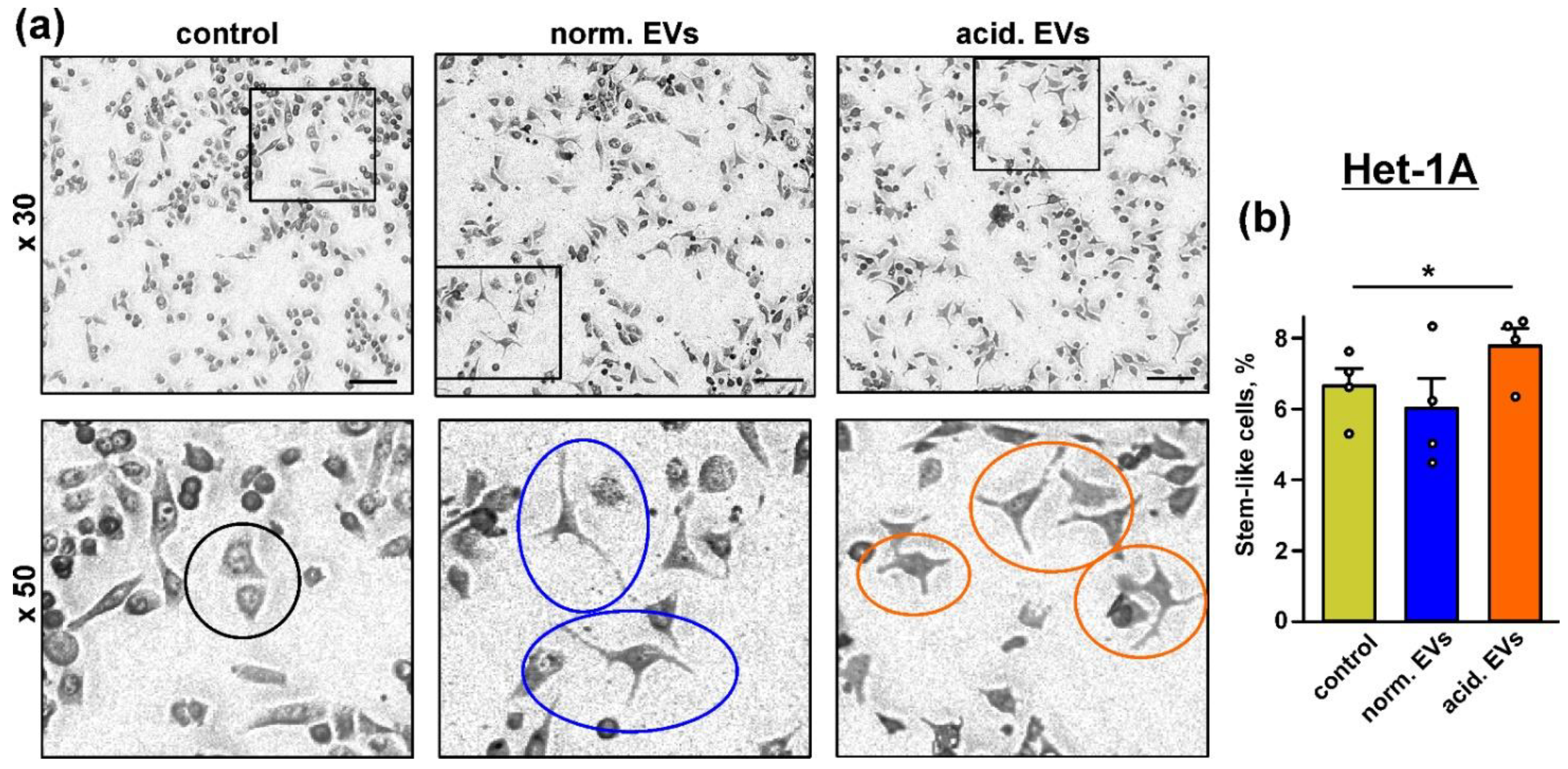
3.3. Metastatic Melanoma EVs Provide “Stem-like” Morphology of Normal Keratinocytes
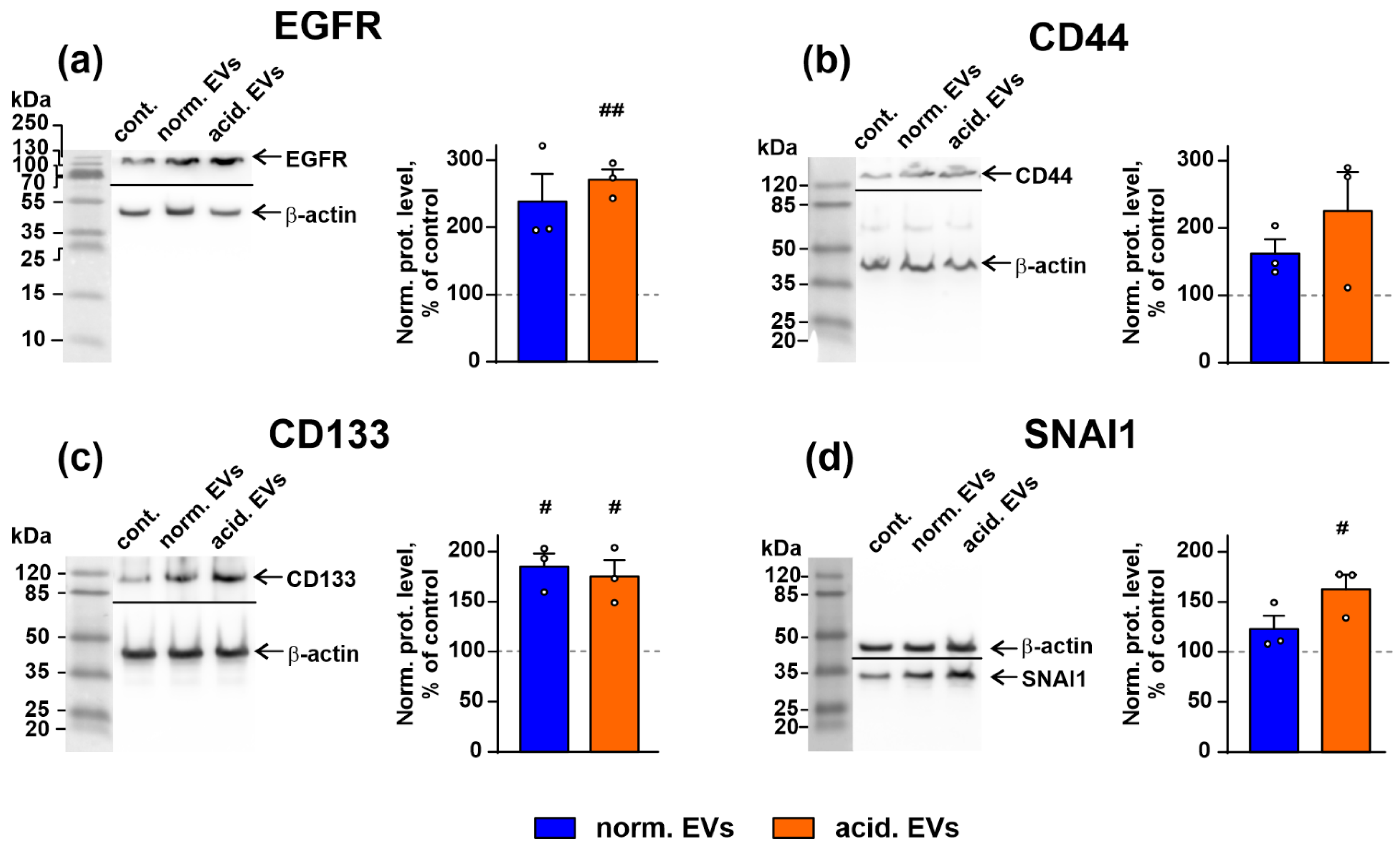
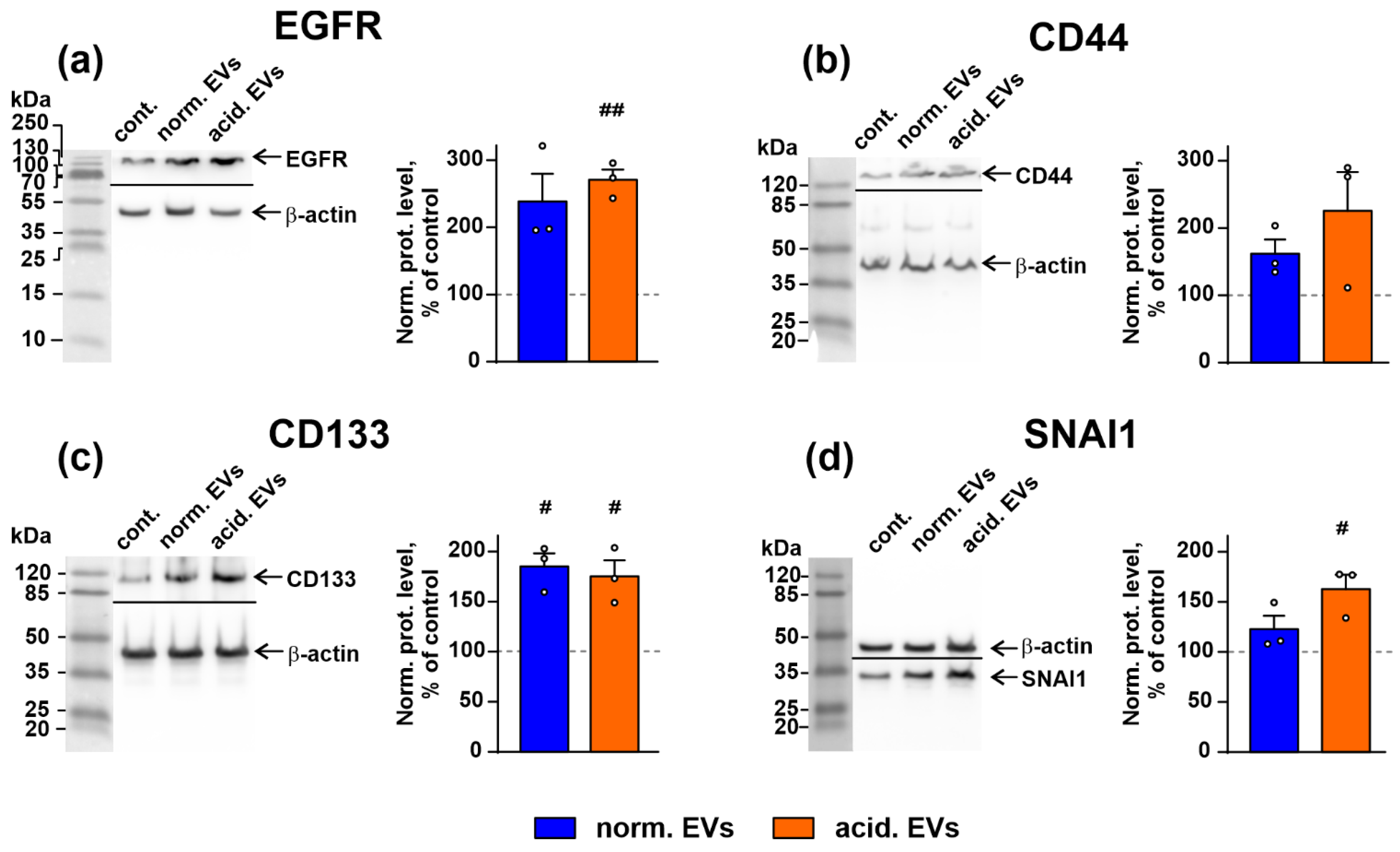
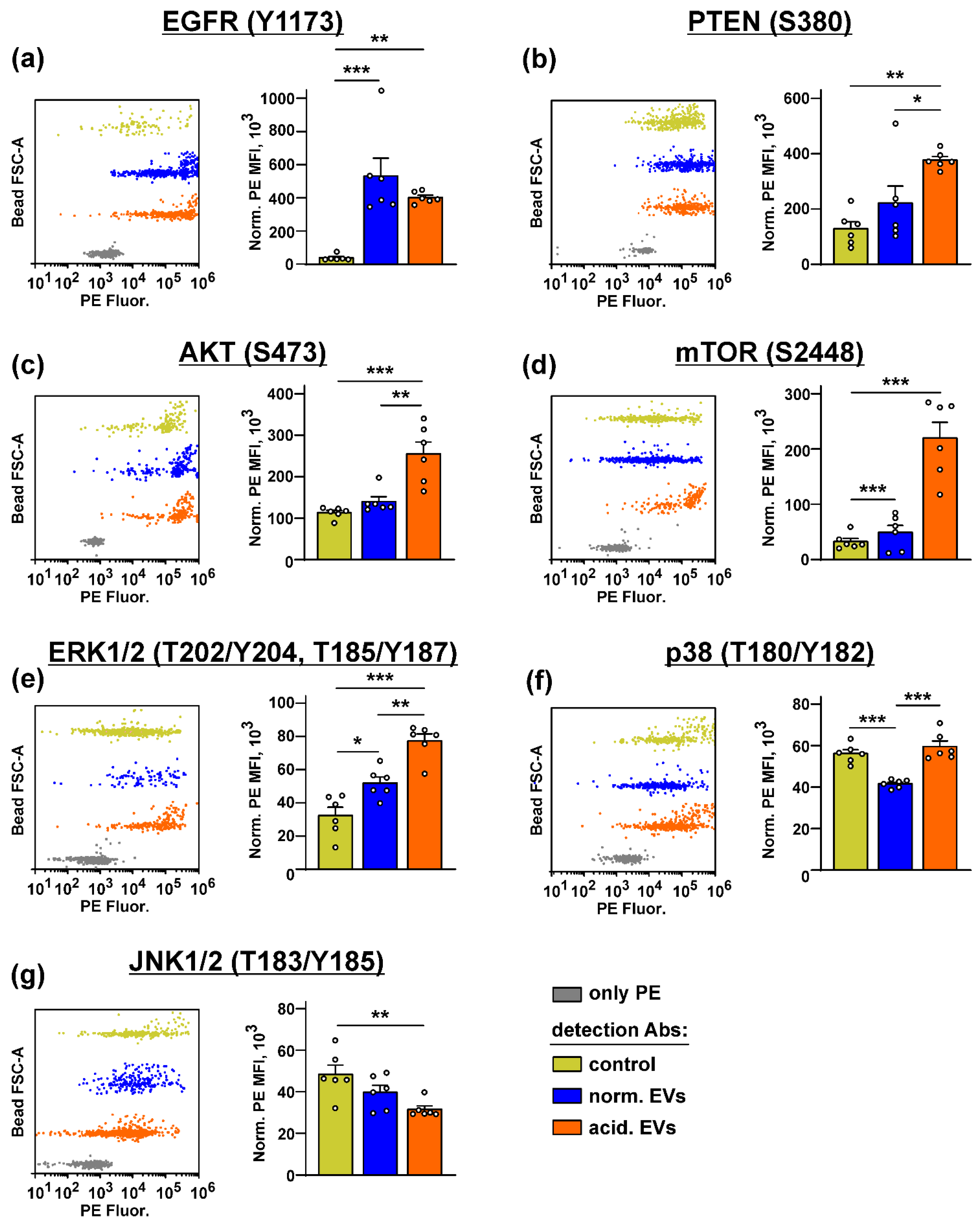
3.4. Metastatic Melanoma EVs Activate Intracellular Signaling Pathways and Factors, Which Mediate Growth, Migration, and Stemness in Normal Keratinocytes
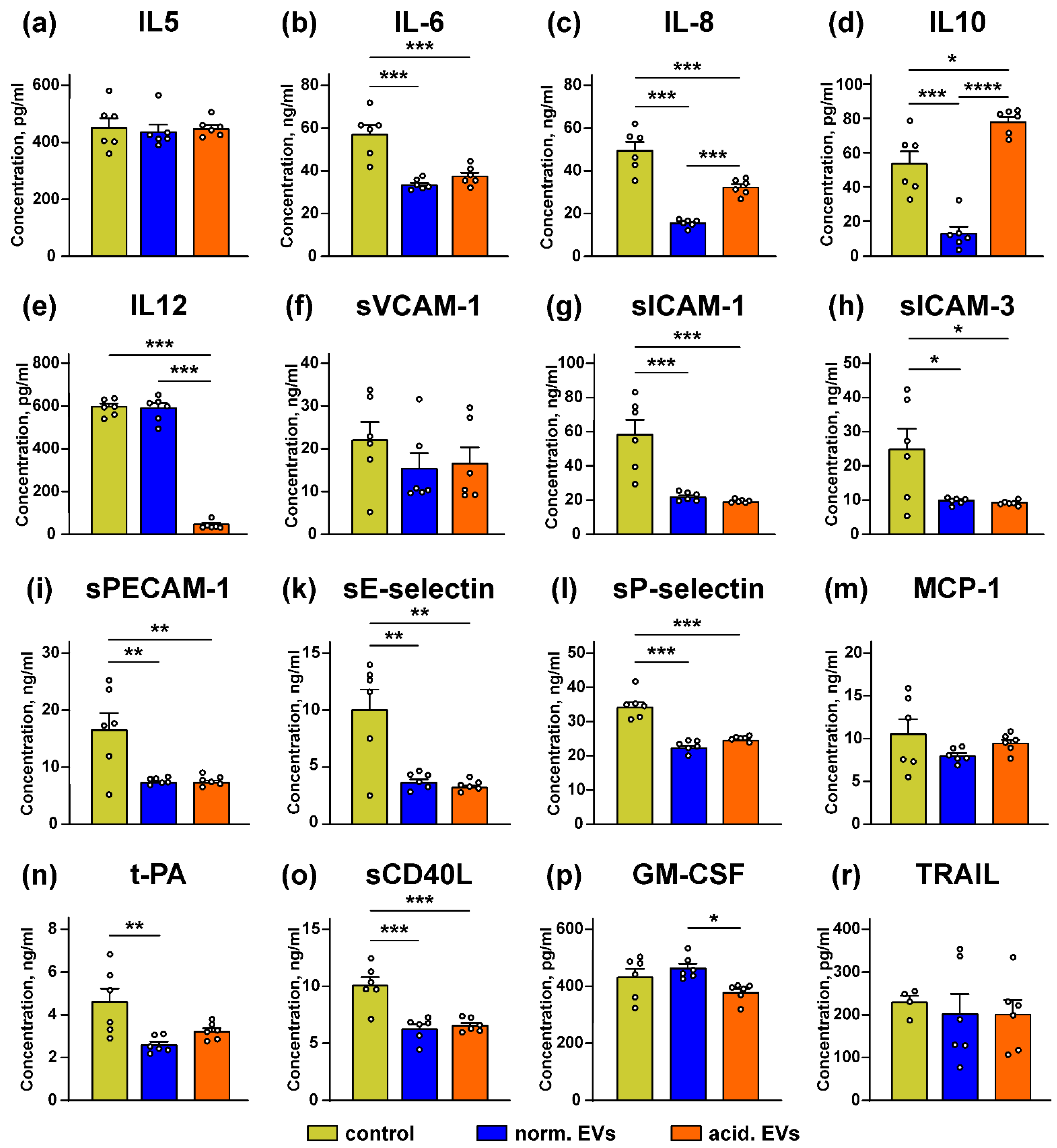
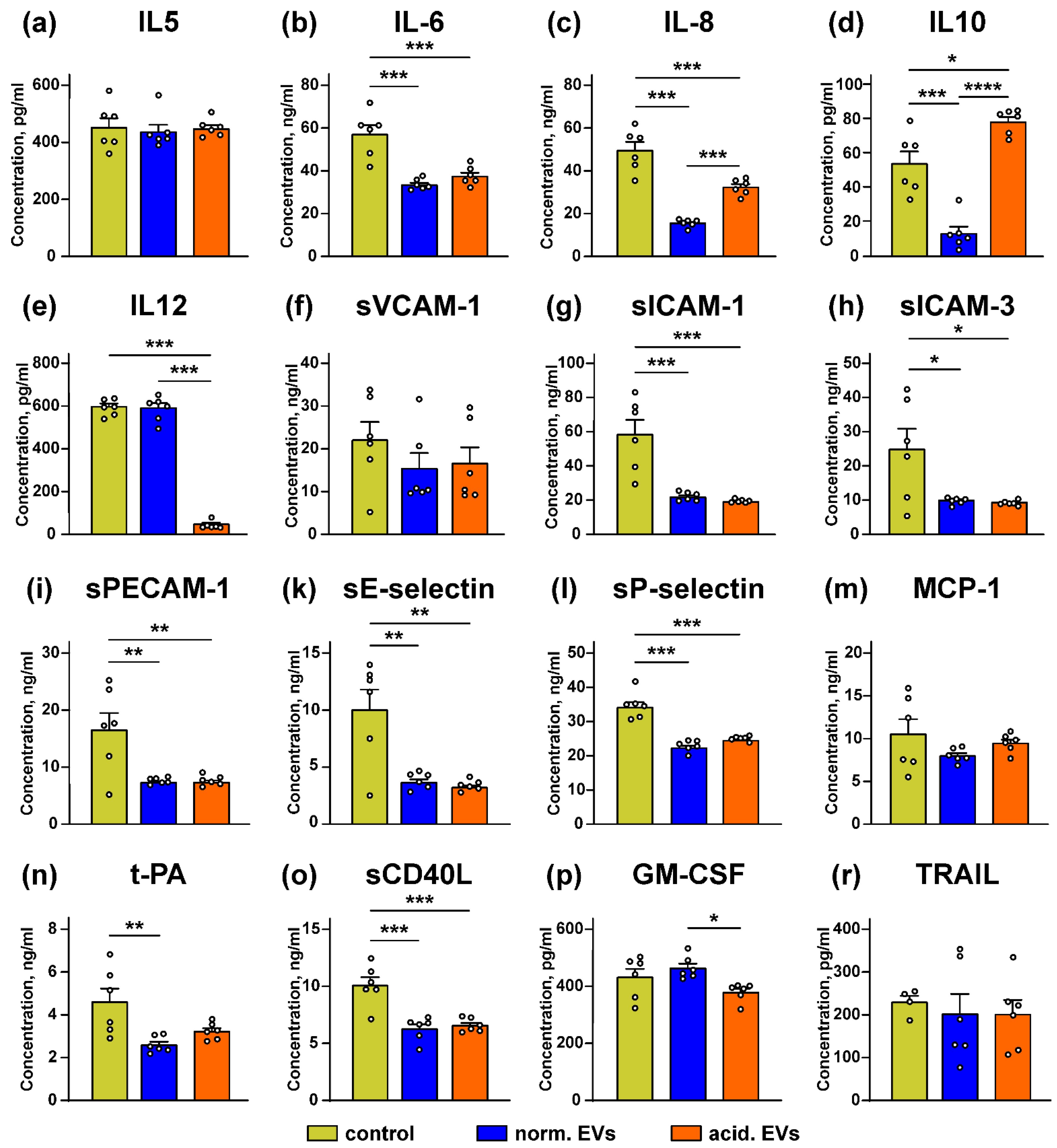
3.5. Metastatic Melanoma EVs Inhibit Release of Immunomodulatory Cytokines and Adhesion Factors by Keratinocytes
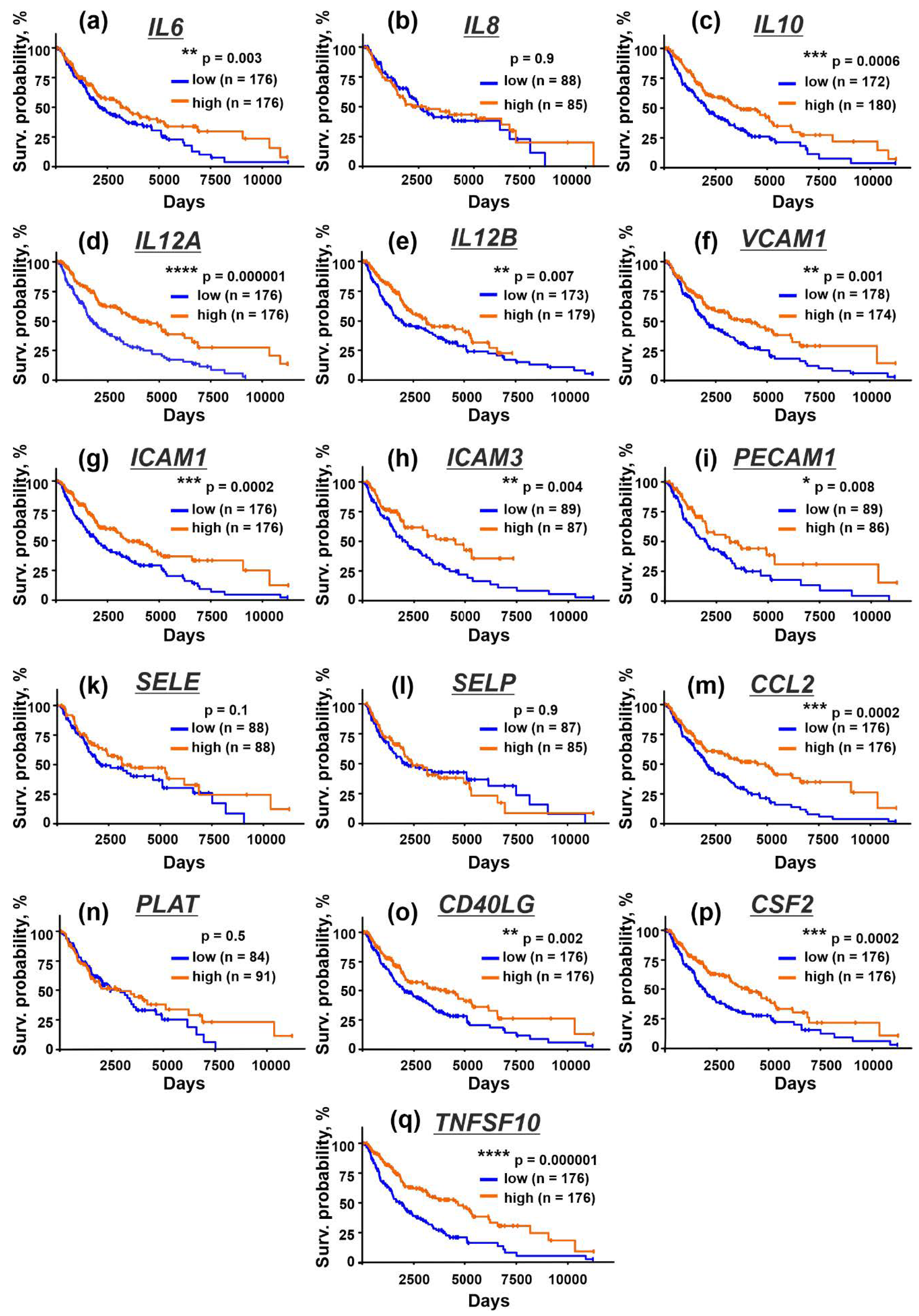
3.6. Upregulation of Immunomodulatory Cytokines and Adhesion Factors in Tissues from Patients with Metastatic Melanoma Correlates with Better Survival Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870. [Google Scholar] [CrossRef] [PubMed]
- Dratkiewicz, E.; Simiczyjew, A.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. Hypoxia and Extracellular Acidification as Drivers of Melanoma Progression and Drug Resistance. Cells 2021, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, M.; Mannavola, F.; Passarelli, A.; Stucci, L.S.; Cives, M.; Silvestris, F. Exosomes in Melanoma: A Role in Tumor Progression, Metastasis and Impaired Immune System Activity. Oncotarget 2018, 9, 20826–20837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.-H.; Cerione, R.A.; Antonyak, M.A. Extracellular Vesicles and Their Roles in Cancer Progression. Methods Mol. Biol. 2021, 2174, 143–170. [Google Scholar] [CrossRef]
- Tickner, J.A.; Urquhart, A.J.; Stephenson, S.-A.; Richard, D.J.; O’Byrne, K.J. Functions and Therapeutic Roles of Exosomes in Cancer. Front. Oncol. 2014, 4, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honegger, A.; Leitz, J.; Bulkescher, J.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Silencing of Human Papillomavirus (HPV) E6/E7 Oncogene Expression Affects Both the Contents and the Amounts of Extracellular Microvesicles Released from HPV-Positive Cancer Cells. Int. J. Cancer 2013, 133, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.K.; Varshney, A.K.; Yadava, P.K. Diversity and Functional Evolution of the Plasminogen Activator System. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Hostetter, D.R.; Moore, S.J.; Winter, M.B. The Multifaceted Roles of Tumor-Associated Proteases and Harnessing Their Activity for Prodrug Activation. Biol. Chem. 2019, 400, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Fernandes, A.R.; Baptista, P.V. Exosome in Tumour Microenvironment: Overview of the Crosstalk between Normal and Cancer Cells. Biomed Res. Int. 2014, 2014, 179486. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, N. Decoding the Secret of Cancer by Means of Extracellular Vesicles. J. Clin. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Weidle, H.U.; Birzele, F.; Kollmorgen, G.; Rüger, R. The Multiple Roles of Exosomes in Metastasis. Cancer Genom. Proteom. 2016, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholia, S.; Ranghino, A.; Garnieri, P.; Lopatina, T.; Deregibus, M.C.; Rispoli, P.; Brizzi, M.F.; Camussi, G. Extracellular Vesicles as New Players in Angiogenesis. Vascul. Pharmacol. 2016, 86, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome Transfer from Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal Lysosomal Trafficking and Enhanced Exosomal Export of Cisplatin in Drug-Resistant Human Ovarian Carcinoma Cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer Cell-Derived Microvesicles Induce Transformation by Transferring Tissue Transglutaminase and Fibronectin to Recipient Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Accelerated Growth of B16BL6 Tumor in Mice through Efficient Uptake of Their Own Exosomes by B16BL6 Cells. Cancer Sci. 2017, 108, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, F.; De Feo, A.; Coscia, C.; Puglisi, R.; Pedini, F.; Pasquini, L.; Bellenghi, M.; Errico, M.C.; Pagani, E.; Carè, A. Exosome-Mediated Transfer of MiR-222 Is Sufficient to Increase Tumor Malignancy in Melanoma. J. Transl. Med. 2016, 14, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells toward a Pro-Metastatic Phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-Serum Amyloid A3-TLR4 Paracrine Cascade Establishes a Pre-Metastatic Phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, J.L. Melanoma Exosomes Enable Tumor Tolerance in Lymph Nodes. Med. Hypotheses 2016, 90, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, J.; Herlyn, M. Melanoma and the Tumor Microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, N.K.; Smalley, K.S.M.; Li, L.; Herlyn, M. Adhesion, Migration and Communication in Melanocytes and Melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef]
- Hachiya, A.; Kobayashi, A.; Yoshida, Y.; Kitahara, T.; Takema, Y.; Imokawa, G. Biphasic Expression of Two Paracrine Melanogenic Cytokines, Stem Cell Factor and Endothelin-1, in Ultraviolet B-Induced Human Melanogenesis. Am. J. Pathol. 2004, 165, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Nasti, T.H.; Timares, L. Inflammasome Activation of IL-1 Family Mediators in Response to Cutaneous Photodamage. Photochem. Photobiol. 2012, 88, 1111–1125. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin Switch from E- to N-Cadherin in Melanoma Progression Is Regulated by the PI3K/PTEN Pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197. [Google Scholar] [CrossRef]
- Böhme, I.; Bosserhoff, A.K. Acidic Tumor Microenvironment in Human Melanoma. Pigment Cell Melanoma Res. 2016, 29, 508–523. [Google Scholar] [CrossRef]
- Bychkov, M.L.; Kirichenko, A.V.; Shulepko, M.A.; Mikhaylova, I.N.; Kirpichnikov, M.P.; Lyukmanova, E.N. Mambalgin-2 Inhibits Growth, Migration, and Invasion of Metastatic Melanoma Cells by Targeting the Channels Containing an ASIC1a Subunit Whose Up-Regulation Correlates with Poor Survival Prognosis. Biomedicines 2021, 9, 1324. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic Microenvironment Plays a Key Role in Human Melanoma Progression through a Sustained Exosome Mediated Transfer of Clinically Relevant Metastatic Molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef] [PubMed]
- Mikhaĭlova, I.N.; Lukashina, M.I.; Baryshnikov, A.I.; Morozova, L.F.; Burova, O.S.; Palkina, T.N.; Kozlov, A.M.; Golubeva, V.A.; Cheremushkin, E.A.; Doroshenko, M.B.; et al. Melanoma cell lines as the basis for antitumor vaccine preparation. Vestn. Ross. Akad. Med. Nauk. 2005, 7, 37–40. [Google Scholar]
- Mikhaylova, I.N.; Kovalevsky, D.A.; Morozova, L.F.; Golubeva, V.A.; Cheremushkin, E.A.; Lukashina, M.I.; Voronina, E.S.; Burova, O.S.; Utyashev, I.A.; Kiselev, S.L.; et al. Cancer/Testis Genes Expression in Human Melanoma Cell Lines. Melanoma Res. 2008, 18, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Hong, C.-S.; Stolz, D.B.; Watkins, S.C.; Whiteside, T.L. Isolation of Biologically-Active Exosomes from Human Plasma. J. Immunol. Methods 2014, 411, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Gong, M.; Hu, Y.; Liu, H.; Zhang, W.; Zhang, M.; Hu, X.; Aubert, D.; Zhu, S.; Wu, L.; et al. Quality and Efficiency Assessment of Six Extracellular Vesicle Isolation Methods by Nano-Flow Cytometry. J. Extracell. Vesicles 2020, 9, 1697028. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.; Bychkov, M.; Sharonov, G.; Efremenko, A.; Shulepko, M.; Kulbatskii, D.; Shenkarev, Z.; Feofanov, A.; Dolgikh, D.; Kirpichnikov, M. Human Secreted Proteins SLURP-1 and SLURP-2 Control the Growth of Epithelial Cancer Cells via Interactions with Nicotinic Acetylcholine Receptors: Actions of Human SLURP Proteins on Cancer Cells. Br. J. Pharmacol. 2018, 175, 1973–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varankar, S.S.; Bapat, S.A. Migratory Metrics of Wound Healing: A Quantification Approach for In Vitro Scratch Assays. Front. Oncol. 2018, 8, 633. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Ohlendorf, J.; Chen, Y.; Taylor, D.D.; Rai, S.N.; Waigel, S.; Zacharias, W.; Hao, H.; McMasters, K.M. Identifying MRNA, MicroRNA and Protein Profiles of Melanoma Exosomes. PLoS ONE 2012, 7, e46874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, N.; Zhou, Q.; Huang, X.; Yu, J.; Han, Q.; Nong, B.; Xiong, Y.; Liang, P.; Li, J.; Feng, M.; et al. Identification of Differentially Expressed Circulating Exosomal LncRNAs in IgA Nephropathy Patients. BMC Immunol. 2020, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- Gennebäck, N.; Hellman, U.; Malm, L.; Larsson, G.; Ronquist, G.; Waldenström, A.; Mörner, S. Growth Factor Stimulation of Cardiomyocytes Induces Changes in the Transcriptional Contents of Secreted Exosomes. J. Extracell. Vesicles 2013, 2, 20167. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.; Jacobs, K.; Ferdinande, L.; Taildeman, J.; Lambert, J.; Peeters, M.; Bracke, M.; Pauwels, P.; Brochez, L. EGFR in Melanoma: Clinical Significance and Potential Therapeutic Target. J. Cutan. Pathol. 2011, 38, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, P.; Neshat, A.; Mokhtari, M.; Rajabi, M.A.; Eftekhari, M.; Tavakoli, P. The Role of VEGF in Melanoma Progression. J. Res. Med. Sci. 2012, 17, 534–539. [Google Scholar] [PubMed]
- Cappello, F.; Conway de Macario, E.; Marasà, L.; Zummo, G.; Macario, A.J.L. Hsp60 Expression, New Locations, Functions and Perspectives for Cancer Diagnosis and Therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Santinon, F.; Flores González, R.E.; del Rincón, S.V. Melanoma Plasticity: Promoter of Metastasis and Resistance to Therapy. Front. Oncol. 2021, 11, 756001. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, Y.; Zhou, J. MiR-31 Functions as an Oncomir Which Promotes Epithelial-Mesenchymal Transition via Regulating BAP1 in Cervical Cancer. Biomed Res. Int. 2017, 2017, 6361420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Liang, H.; Wang, Y.; Zhang, W.; Zhou, Y.; Chen, S.A.; Yu, M.; Cui, S.; Liu, M.; Wang, N.; et al. MiR-96 Promotes Cell Proliferation, Migration and Invasion by Targeting PTPN9 in Breast Cancer. Sci. Rep. 2016, 6, 37421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Ma, P.; Wu, D.; Shu, Y.; Gao, W. Functions and Mechanisms of MicroRNA-31 in Human Cancers. Biomed. Pharmacother. 2018, 108, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ishak Gabra, M.B.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Li, H.; Milman, N.; Liu, J.; Reid, M.A.; Locasale, J.W.; et al. MiR-135 Suppresses Glycolysis and Promotes Pancreatic Cancer Cell Adaptation to Metabolic Stress by Targeting Phosphofructokinase-1. Nat. Commun. 2019, 10, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. MiR221/222 in Cancer: Their Role in Tumor Progression and Response to Therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 Promotes Cell Transformation by Targeting the Programmed Cell Death 4 Gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Handorf, C.R.; Pfeffer, L.M. MicroRNA MiR-21 Regulates the Metastatic Behavior of B16 Melanoma Cells. J. Biol. Chem. 2011, 286, 39172–39178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wang, Q.; Zhu, X.-H. MiR-135b Is a Novel Oncogenic Factor in Cutaneous Melanoma by Targeting LATS2. Melanoma Res. 2019, 29, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, F.; Errico, M.C.; Bottero, L.; Segnalini, P.; Biffoni, M.; Felli, N.; Mattia, G.; Petrini, M.; Colombo, M.P.; Carè, A. MicroRNA-221/-222 Pathway Controls Melanoma Progression. Eur. J. Cancer Suppl. 2008, 6, 122. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. MiR-7 Reverses the Resistance to BRAFi in Melanoma by Targeting EGFR/IGF-1R/CRAF and Inhibiting the MAPK and PI3K/AKT Signaling Pathways. Oncotarget 2016, 7, 53558–53570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunel, N.O.; Martin, M.T. When the Search for Stemness Genes Meets the Skin Substitute Bioengineering Field: KLF4 Transcription Factor under the Light. Cells 2020, 9, 2188. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Konishi, Y.; Kosaka, N.; Katsuda, T.; Kato, T.; Ochiya, T. Comparative Marker Analysis of Extracellular Vesicles in Different Human Cancer Types. J. Extracell. Vesicles 2013, 2, 20424. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Bianchini, F.; Torre, E.; Calorini, L. Contribution of Acidic Melanoma Cells Undergoing Epithelial-to-Mesenchymal Transition to Aggressiveness of Non-Acidic Melanoma Cells. Clin. Exp. Metastasis 2014, 31, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Marck, V.L.V.; Bracke, M.E. Epithelial-Mesenchymal Transitions in Human Cancer; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Yu, F.; Li, J.; Chen, H.; Fu, J.; Ray, S.; Huang, S.; Zheng, H.; Ai, W. Kruppel-like Factor 4 (KLF4) Is Required for Maintenance of Breast Cancer Stem Cells and for Cell Migration and Invasion. Oncogene 2011, 30, 2161–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The Role of Pluripotency Factors to Drive Stemness in Gastrointestinal Cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, M.M.; Palacios-Helgeson, L.K.; Wah, L.S.; Jackson, T.A. Rapid Estrogen Signaling Negatively Regulates PTEN Activity through Phosphorylation in Endometrial Cancer Cells. Horm. Cancer 2014, 5, 218–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Zhou, C.; Bi, Z.; Wan, Y. EGF-Induced Cell Migration Is Mediated by ERK and PI3K/AKT Pathways in Cultured Human Lens Epithelial Cells. J. Ocul. Pharmacol. Ther. 2006, 22, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Wu, S. MiR-451: A Novel Biomarker and Potential Therapeutic Target for Cancer. Onco Targets Ther. 2019, 12, 11069–11082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, S.; Mori, T.; Otsuka, Y.; Yamada, N.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Maruo, K.; Akao, Y. Anti-Oncogenic MicroRNA-203 Induces Senescence by Targeting E2F3 Protein in Human Melanoma Cells. J. Biol. Chem. 2012, 287, 11769–11777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw-Denboba, A.; Balestrieri, E.; Serafino, A.; Cipriani, C.; Bucci, I.; Sorrentino, R.; Sciamanna, I.; Gambacurta, A.; Sinibaldi-Vallebona, P.; Matteucci, C. HERV-K Activation Is Strictly Required to Sustain CD133+ Melanoma Cells with Stemness Features. J. Exp. Clin. Cancer Res. 2017, 36, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and BHLH Factors in Tumour Progression: An Alliance against the Epithelial Phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Ramez, M.; Gilad, E.; Singleton, P.A.; Man, M.-Q.; Crumrine, D.A.; Elias, P.M.; Feingold, K.R. Hyaluronan-CD44 Interaction Stimulates Keratinocyte Differentiation, Lamellar Body Formation/Secretion, and Permeability Barrier Homeostasis. J. Investig. Dermatol. 2006, 126, 1356–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.F.; Pöyry, T.A.A.; Stoneley, M.; Willis, A.E. Signaling from MTOR to EIF2α Mediates Cell Migration in Response to the Chemotherapeutic Doxorubicin. Sci. Signal. 2019, 12, eaaw6763. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Okada, Y.; Miyamoto, T.; Yamanaka, O.; Ohnishi, Y.; Ooshima, A.; Liu, C.-Y.; Weng, D.; Kao, W.W.-Y. Role of P38 MAP Kinase in Regulation of Cell Migration and Proliferation in Healing Corneal Epithelium. Investig. Ophthalmol. Vis. Sci. 2004, 45, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-Signaling: A Multiplexing Hub in Programmed Cell Death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK Dual Inhibitors: The Way Forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, F.C.; Giles, K.M.; Candy, P.A.; Ali, A.; Ganda, C.; Epis, M.R.; Webster, R.J.; Leedman, P.J. Regulation of Epidermal Growth Factor Receptor Signaling and Erlotinib Sensitivity in Head and Neck Cancer Cells by MiR-7. PLoS ONE 2012, 7, e47067. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, E22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandoga, A.; Kessler, J.S.; Meissner, H.; Hanschen, M.; Corada, M.; Motoike, T.; Enders, G.; Dejana, E.; Krombach, F. Junctional Adhesion Molecule-A Deficiency Increases Hepatic Ischemia-Reperfusion Injury despite Reduction of Neutrophil Transendothelial Migration. Blood 2005, 106, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, M.; Viñas, O.; Pino-Otín, M.R.; Places, L.; Martínez-Cáceres, E.; Barceló, J.J.; Miralles, A.; Vilella, R.; de la Fuente, M.A.; Vives, J. CD50 (Intercellular Adhesion Molecule 3) Stimulation Induces Calcium Mobilization and Tyrosine Phosphorylation through P59fyn and P56lck in Jurkat T Cell Line. J. Exp. Med. 1994, 179, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.R.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; van Damme, J.; Mantovani, A. Autocrine Production of IL-10 Mediates Defective IL-12 Production and NF-Kappa B Activation in Tumor-Associated Macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Bhurani, V.; Dalai, S.K. Therapeutic Potentials of IL-10 versus IL-12; IntechOpen: London, UK, 2018; ISBN 978-1-78923-509-8. [Google Scholar]
- Liu, J.; Zhang, H.; Liu, Y.; Wang, K.; Feng, Y.; Liu, M.; Xiao, X. KLF4 Regulates the Expression of Interleukin-10 in RAW264.7 Macrophages. Biochem. Biophys. Res. Commun. 2007, 362, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (UPA) and Its Receptor (UPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chonov, D.C.; Ignatova, M.M.K.; Ananiev, J.R.; Gulubova, M.V. IL-6 Activities in the Tumour Microenvironment. Part 1. Open Access Maced. J. Med. Sci. 2019, 7, 2391–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, M.; O’Garra, A. The Regulation of IL-10 Production by Immune Cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, M.; Eto, M.; Young, G.D.; Berd, D.; Mastrangelo, M.J.; Tamura, Y.; Harigaya, K.; Sato, T. Interleukin 6 Mediates Production of Interleukin 10 in Metastatic Melanoma. Cancer Immunol. Immunother. 2012, 61, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.-C.; et al. IL-6 Trans-Signaling Licenses Mouse and Human Tumor Microvascular Gateways for Trafficking of Cytotoxic T Cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming Anti-Tumor Immunity by Radiotherapy: Dying Tumor Cell-Derived DAMPs Trigger Endothelial Cell Activation and Recruitment of Myeloid Cells. Oncoimmunology 2018, 8, e1523097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Kapoor, P.; Spongberg, E.; Davis, D.; Vorwald, V.M.; Couts, K.L.; Gao, D.; Smith, D.E.; Borgers, J.S.W.; et al. IL-6 and IL-8 Are Linked With Myeloid-Derived Suppressor Cell Accumulation and Correlate With Poor Clinical Outcomes in Melanoma Patients. Front. Oncol. 2019, 9, 1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P.; Stow, J.L. Cytokine Release from Innate Immune Cells: Association with Diverse Membrane Trafficking Pathways. Blood 2011, 118, 9–18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | “Normal” EVs (pH 7.4) | “Acidic” EVs (pH 6.5) |
|---|---|---|
| EGFR | - | 1.72 × 10−5 ± 4.7 × 10−6 |
| PDGFRA | - | - |
| TNFA | - | - |
| BDNF | - | - |
| VEGFA | - | 8.49 × 10−4 ± 1.7 × 10−4 |
| ITGA2 | - | - |
| ITGA3 | - | 2.87 × 10−3 ± 6.4 × 10−4 |
| ITGV | - | 1.1 × 10−5 ± 5.5 × 10−6 |
| HSP60 | - | 2.26 × 10−6 ± 1.7 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bychkov, M.L.; Kirichenko, A.V.; Mikhaylova, I.N.; Paramonov, A.S.; Yastremsky, E.V.; Kirpichnikov, M.P.; Shulepko, M.A.; Lyukmanova, E.N. Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes. Biomedicines 2022, 10, 660. https://doi.org/10.3390/biomedicines10030660
Bychkov ML, Kirichenko AV, Mikhaylova IN, Paramonov AS, Yastremsky EV, Kirpichnikov MP, Shulepko MA, Lyukmanova EN. Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes. Biomedicines. 2022; 10(3):660. https://doi.org/10.3390/biomedicines10030660
Chicago/Turabian StyleBychkov, Maxim L., Artem V. Kirichenko, Irina N. Mikhaylova, Alexander S. Paramonov, Evgeny V. Yastremsky, Mikhail P. Kirpichnikov, Mikhail A. Shulepko, and Ekaterina N. Lyukmanova. 2022. "Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes" Biomedicines 10, no. 3: 660. https://doi.org/10.3390/biomedicines10030660
APA StyleBychkov, M. L., Kirichenko, A. V., Mikhaylova, I. N., Paramonov, A. S., Yastremsky, E. V., Kirpichnikov, M. P., Shulepko, M. A., & Lyukmanova, E. N. (2022). Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes. Biomedicines, 10(3), 660. https://doi.org/10.3390/biomedicines10030660