
Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery


 ,
, 

Abstract
1. Introduction
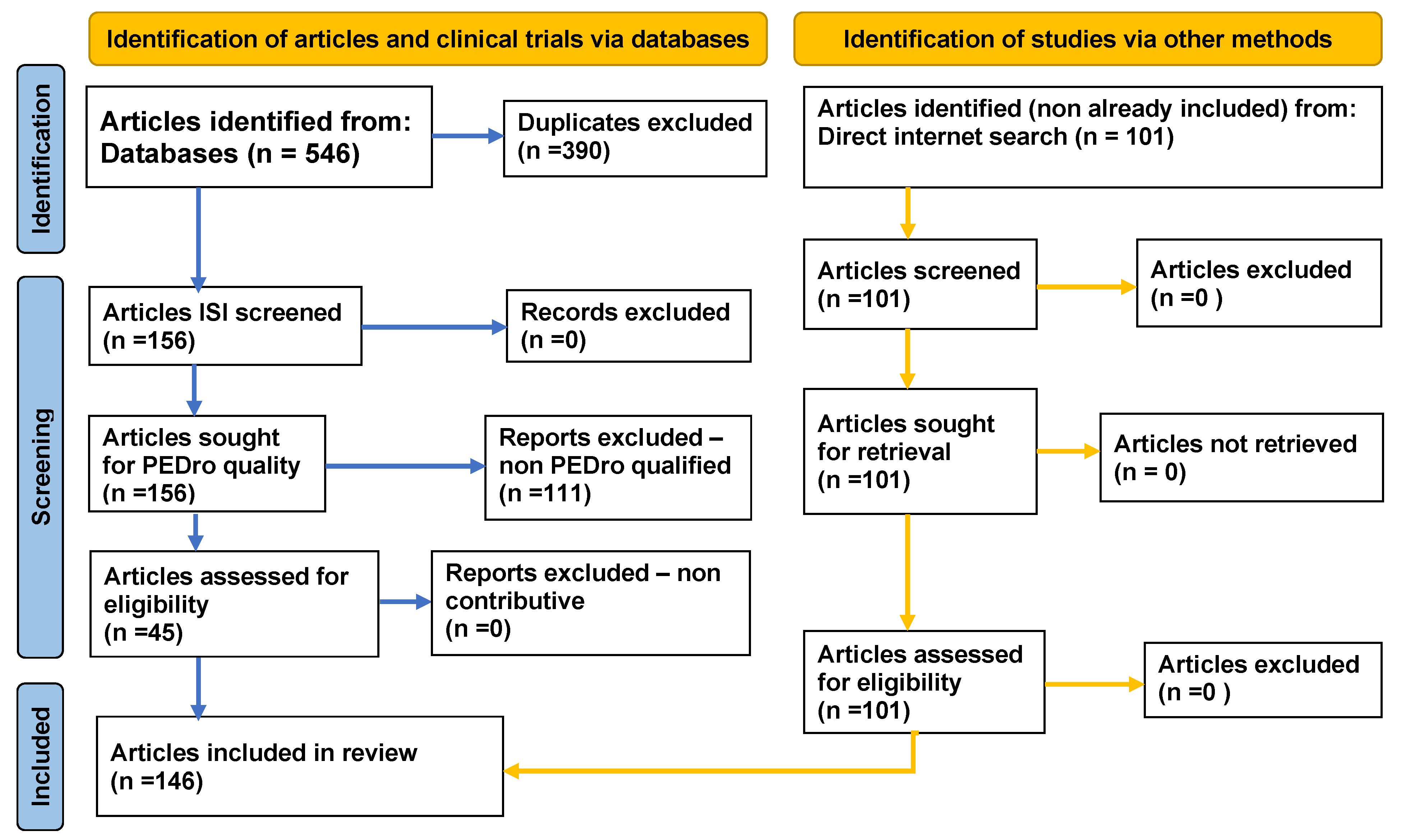
2. Method
3. Results
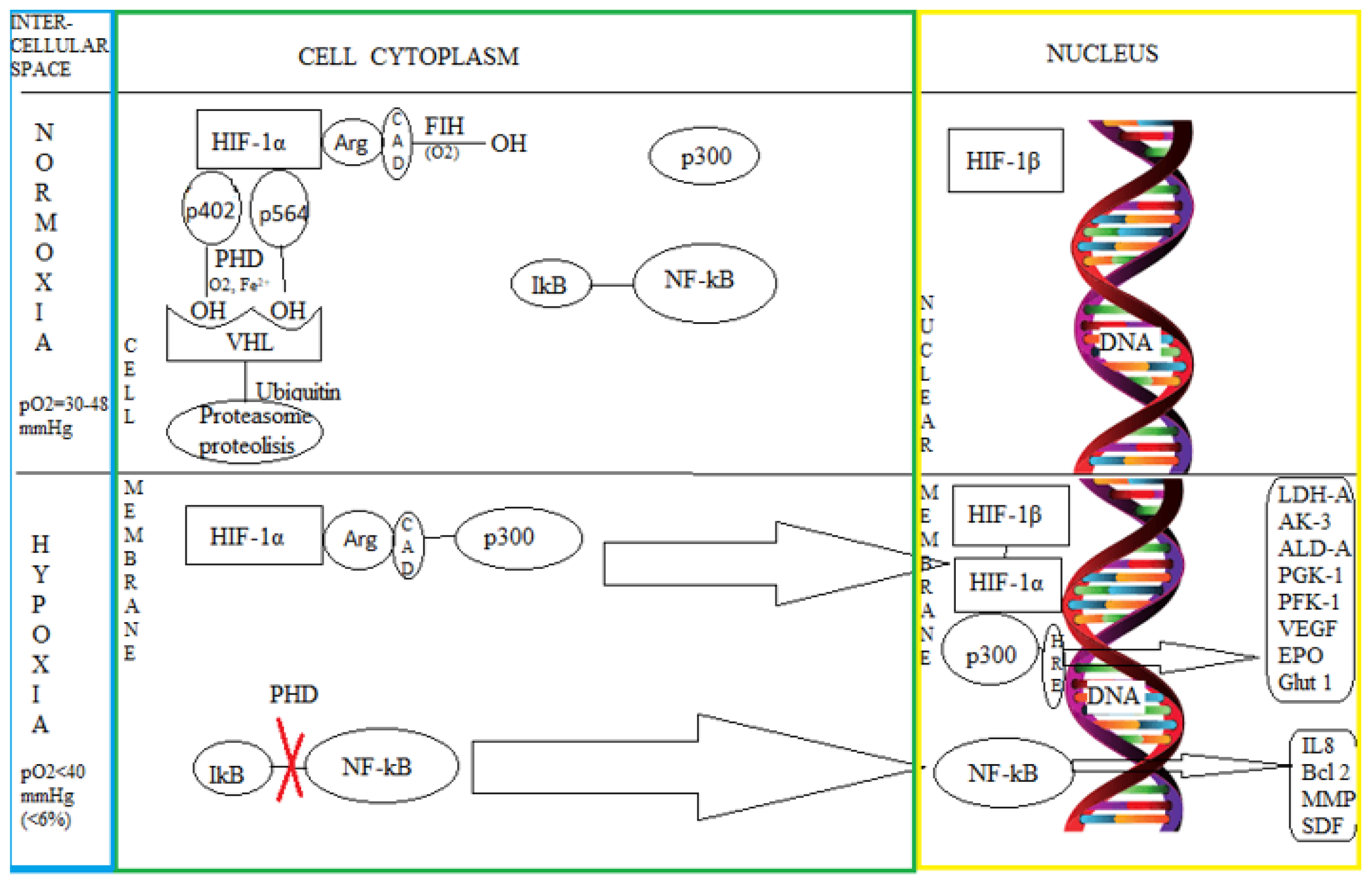
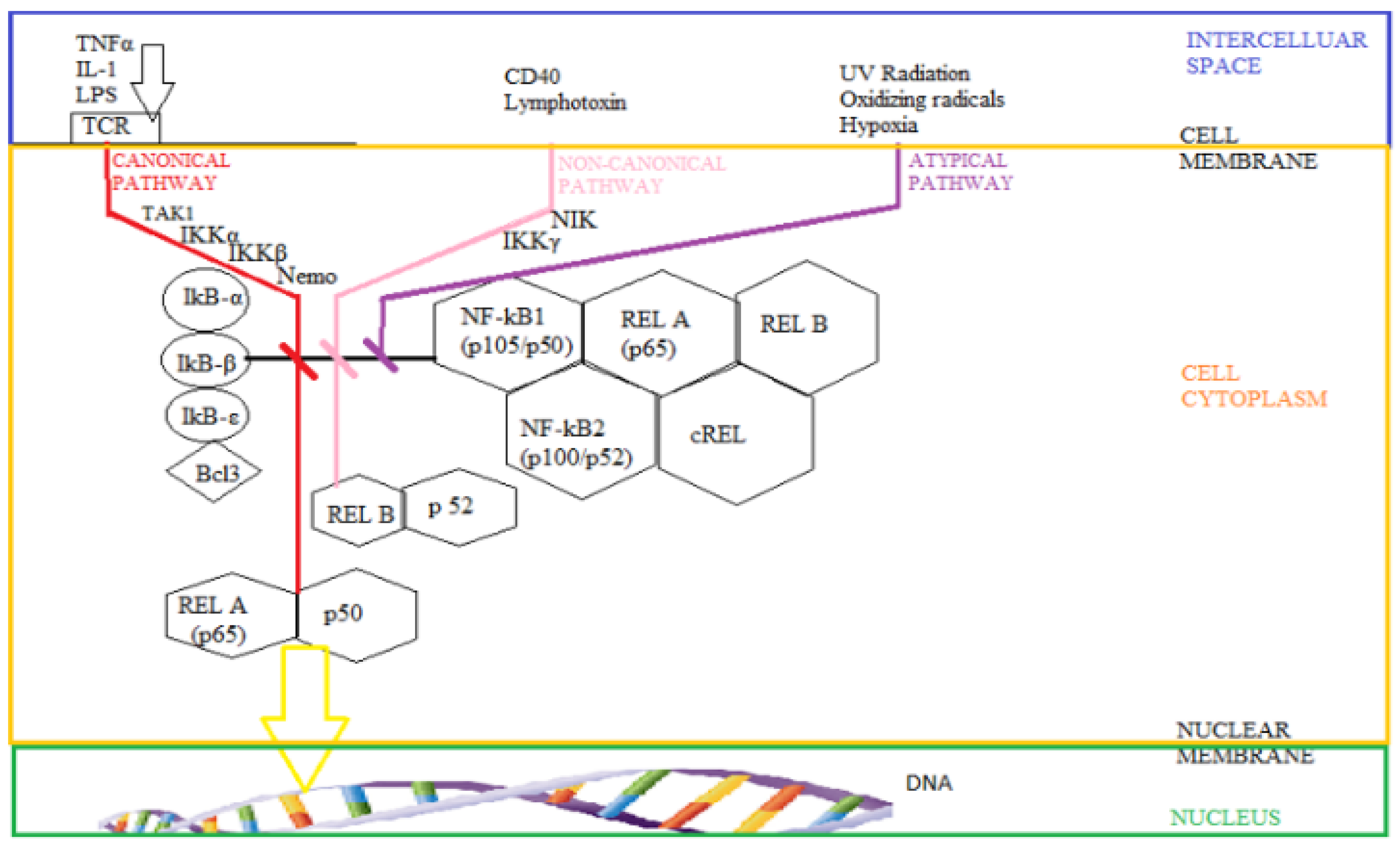
3.1. The Intimate Mechanisms of Hypoxia
3.2. The Influence of Hypoxia on the Nervous Tissue
3.3. Newborn Hypoxic-Ischemic Encephalopathy
3.4. Adult Brain Ischemic Vascular Lesions
3.5. Adult Spinal Cord Injury and Hypoxia
3.6. Hypoxia and Functional Recovery

{kind=link}
{kind=link}
{kind=link}
| ||
| Article | Ref. no | Subject |
| (Thornton, 2017) | [11] | Hypoxic-ischemic lesions cause energy disorders in cell metabolism, leading to cell death through apoptosis, necrosis and autolysis |
| (Cai, 2019) | [14] | MCAO mice showed an invasion of immune cells into the brain |
| (Nowak-Sliwinskaet, 2018) | [22] | HIF-1 is essential for normal development and the response to ischemia/hypoxia, tumor development, energy metabolism, angiogenesis, apoptosis, proliferation, and vasomotor function |
| (Yuniati, 2019) | [43] | NF-kB modulates the expression of numerous proteins |
| (Gschwandtner, 2019) | [44] | apoptosis and the inhibition of programmed cell death |
| (Yang, 2017) | [47] | NF-kB increases the expression of IL-8, inducing angiogenesis that contributes to the generation of neovascularization in hypoxia |
| ||
| (Clark, 2019) | [54] | Nerve tissue is made up of neurons and glial cells |
| (Miller, 2017) | [55] | Microglia are derived from erythromyeloid progenitors |
| (Greenhalgh, 2018) | [56] | Microglia have an essential role |
| (Barrett, 2017) | [57] | cerebral homeostasis |
| (Ginwala, 2019) | [58] | NO synthase and NF-κB activation |
| (Liu, 2019) | [59] | Melatonin is a pineal hormone with anti-inflammatory effect |
| (Becerra-Calixto, 2017) | [60] | calcium and potassium homeostasis |
| (Islinger, 2018) | [61] | HIF1α stimulates the production of peroxisomes |
| (Gorgulho, 2019) | [63] | High mobility group box 1 (HMGB1) protein |
| (Kim, 2017) | [64] | neuroinflammatory response, pathogenesis of ischemic stroke |
| ||
| (Geisler, 2019) | [75] | reduced blood flow and brain oxygenation |
| (Rohowetz, 2018) | [76] | Mitochondria tend to hyperpermeabilize |
| (Weiskirchen, 2016) | [77] | ROS are involved in cell physiological / pathological processes, |
| (de Faria, 2019) | [80] | phagocytosis processes of cell debris |
| ||
| (Carvajal, 2016) | [85] | The ionotropic glutamate receptor AMPA |
| (Galicia-Garcia, 2020) | [86] | Stroke (neurological condition) - individuals, family and social |
| (Pennisi, 2020) | [87] | Stroke neurological condition and SARS-CoV-2 |
| (Shahabipour, 2017) | [88] | Aβ proteins along with the decreased expression of neprilysin |
| (Tanaka, 2020) | [90] | Aβ proteins - Alzheimer's dementia |
| (Şekerdağ, 2018) | [91] | acute phase post stroke VEGF increases permeability of BBB |
| (Morya, 2019) | [92] | Primary and secondary lesions occur in traumatic brain injury |
| (Ramirez, 2018) | [94] | proliferation, signal transduction |
| (Iraci, 2016) | [95] | regulation, miRNA - traumatic brain injury |
| (Ciregia, 2017) | [96] | Traumatic brain injury (TBI) biomarker |
| ||
| (Poniatowski, 2017) | [98] | risk of polytrauma |
| (Lin, 2020) | [99] | primary and secondary lesions |
| (Kim, 2019) | [102] | Overexpression of the GM-CSF gene protects |
| ||
| (Miranda, 2019) | [111] | intermittent exposure to hypoxia |
| (Zhou, 2016) | [117] | hypoxic stimulation is thought to stimulate neuroplasticity |
| (Ke, 2019) | [119] | intermittent hypoxia may induce HIF-1α expression |
| (Tan, 2018) | [124] | VEGF production is stimulated by neuropeptide Y (NPY) |
| (Yung, 2020) | [126] | NPY is an orexigenic hormone, negatively regulated by insulin |
| (Gaforio, 2019) | [130] | Mediterranean diet based on an abundant consumption of olive oil |
| (Angeloni, 2017) | [131] | olive oil has anti-inflammatory and immunomodulatory effects |
| (Libro, 2016) | [132] | Natural flavonoids (wogonin, curcumin, apigenin, quercetin) |
| (Teleanu, 2019) | [133] | anti-inflammatory effect |
| (Ilyasov, 2018) | [134] | inhibit the production of IL-6, TNF-α, and IL-1β - MAPK pathway |
| (Gu, 2020) | [137] | apamin (bee venom) |
| (Cramer, 2020) | [138] | cannabinoid receptor agonists on oligodendrocytes |
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through HIFs and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Hadanny, A.; Efrati, S. The Hyperoxic-Hypoxic Paradox. Biomolecules 2020, 10, 958. [Google Scholar] [CrossRef] [PubMed]
- Hoteteu, M.; Munteanu, C.; Ionescu, E.V.; Almășan, R.E. Bioactive substances of the Techirghiol therapeutic mud. Balneo Res. J. 2018, 9, 5–10. [Google Scholar] [CrossRef]
- Yang, L.; Roberts, D.; Takhar, M.; Erho, N.; Bibby, B.A.S.; Thiruthaneeswaran, N.; Bhandari, V.; Cheng, W.C.; Haider, S.; McCorry, A.M.B.; et al. Development and Validation of a 28-gene Hypoxia-related Prognostic Signature for Localized Prostate Cancer. EBioMedicine 2018, 31, 182–189. [Google Scholar] [CrossRef]
- Navarrete-Opazo, A.; Mitchell, G.S. Therapeutic potential of intermittent hypoxia: A matter of dose. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1181–R1197. [Google Scholar] [CrossRef]
- Wu, D.; Yotnda, P. Induction and testing of hypoxia in cell culture. J. Vis. Exp. 2011, 54, e2899. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Joshi, S.; Ornstein, E.; Young, W.L. Cerebral and Spinal Cord Blood Flow. In Cottrell and Young’s Neuroanesthesia, 5th ed.; Mosby: Maryland Heights, MI, USA, 2010; pp. 17–59. ISBN 9780323059084. [Google Scholar] [CrossRef]
- Marcus, M.L.; Heistad, D.D.; Ehrhardt, J.C.; Abboud, F.M. Regulation of total and regional spinal cord blood flow. Circ. Res. 1977, 41, 128–134. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Thornton, C.; Leaw, B.; Mallard, C.; Nair, S.; Jinnai, M.; Hagberg, H. Cell death in the developing brain after hypoxia-ischemia. Front. Cell. Neurosci. 2017, 11, 1–19. [Google Scholar] [CrossRef]
- Zhaoa, L.-R.; Willing, A. Enhancing endogenous capacity to repair a stroke-damaged brain: An evolving field for stroke research. Prog. Neurobiol. 2018, 163, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, D.S.; Dubrovskaya, N.M.; Tumanova, N.L.; Zhuravin, I.A. Prenatal hypoxia in different periods of embryogenesis differentially affects cell migration, neuronal plasticity, and rat behavior in postnatal ontogenesis. Front. Neurosci. 2016, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Pan, C.; Ghasemigharagoz, A.; Todorov, M.I.; Förstera, B.; Zhao, S.; Bhatia, H.S.; Parra-Damas, A.; Mrowka, L.; Theodorou, D.; et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull-meninges connections. Nat. Neurosci. 2019, 22, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kerridge, C.; Kozlova, D.I.; Nalivaeva, N.N.; Turner, A.J. Hypoxia affects neprilysin expression through caspase activation and an APP intracellular domain-dependent mechanism. Front. Neurosci. 2015, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, R.; Jankovic, J. Movement disorders in cerebrovascular disease. Lancet Neurol. 2013, 12, 597–608. [Google Scholar] [CrossRef]
- Onose, G.; Anghelescu, A.; Blendea, D.; Ciobanu, V.; Daia, C.; Firan, F.C.; Oprea, M.; Spinu, A.; Popescu, C.; Ionescu, A.; et al. Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 907. [Google Scholar] [CrossRef]
- Onose, G.; Anghelescu, A.; Blendea, C.D.; Ciobanu, V.; Daia, C.O.; Firan, F.C.; Munteanu, C.; Oprea, M.; Spinu, A.; Popescu, C. Non-invasive, non-pharmacological/bio-technological interventions towards neurorestoration upshot after ischemic stroke, in adults—systematic, synthetic, literature review. Front. Biosci. 2021, 26, 1204–1239. [Google Scholar] [CrossRef]
- Lehotskỳ, J.; Tothová, B.; Kovalská, M.; Dobrota, D.; Benová, A.; Kalenská, D.; Kaplán, P. Role of homocysteine in the ischemic stroke and development of ischemic tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef]
- Pang, H.; Fu, Q.; Cao, Q.; Hao, L.; Zong, Z. Sex differences in risk factors for stroke in patients with hypertension and hyperhomocysteinemia. Sci. Rep. 2019, 9, 14313. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Sköld, M.K.; Marti, H.H.; Lindholm, T.; Lindå, H.; Hammarberg, H.; Risling, M.; Cullheim, S. Induction of HIF1α but not HIF2α in motoneurons after ventral funiculus axotomy—Implication in neuronal survival strategies. Exp. Neurol. 2004, 188, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S. Ratcliffe1999. Nature 1999, 459, 271–275. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Ortiz-Prado, E.; Dunn, J.F.; Vasconez, J.; Castillo, D.; Viscor, G. Partial pressure of oxygen in the human body: A general review. Am. J. Blood Res. 2019, 9, 1–14. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30899601%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6420699 (accessed on 28 November 2021). [PubMed]
- Burslem, G.M.; Kyle, H.F.; Nelson, A.; Edwards, T.A.; Wilson, A.J. Hypoxia inducible factor (HIF) as a model for studying inhibition of protein-protein interactions. Chem. Sci. 2017, 8, 4188–4202. [Google Scholar] [CrossRef] [PubMed]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1α, HIF-2α, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef]
- Appelhoffl, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef]
- Hao, J.; Chen, X.; Fu, T.; Liu, J.; Yu, M.; Han, W.; He, S.; Qian, R.; Zhang, F. The Expression of VHL (Von Hippel-Lindau) After Traumatic Spinal Cord Injury and Its Role in Neuronal Apoptosis. Neurochem. Res. 2016, 41, 2391–2400. [Google Scholar] [CrossRef]
- Chen, F.; Qi, Z.; Luo, Y.; Taylor Hinchliffe, G.D.; Xia, Y.; Ji, X. Non-pharmaceutical therapies for stroke: Mechanisms and clinical implications. Prog Neurobiol. 2014, 246–269. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial-specific regulation of hypoxiainducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Kayama, F.; Oguma, E.; Willmore, W.G.; Hradecky, P.; Bunn, H.F. Cadmium and platinum suppression of erythropoietin production in cell culture: Clinical implications. Blood 2000, 96, 3743–3747. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.L.; Sager, T.N.; Lotharius, J.; Witten, L.; Mørk, A.; Egebjerg, J.; Thirstrup, K. HIF prolyl hydroxylase inhibition increases cell viability and potentiates dopamine release in dopaminergic cells. J. Neurochem. 2010, 115, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.A.K.; Bainbridge, J.W.B. Oxygen sensing in retinal health and disease. Ophthalmologica 2012, 227, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- D’ignazio, L.; Rocha, S. Hypoxia induced NF-kB. Cells 2016, 5, 10. [Google Scholar] [CrossRef]
- Simmons, L.J.; Surles-Zeigler, M.C.; Li, Y.; Ford, G.D.; Newman, G.D.; Ford, B.D. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J. Neuroinflamma. 2016, 13, 237. [Google Scholar] [CrossRef]
- Osipo, C.; Golde, T.E.; Osborne, B.A.; Miele, L.A. Off the beaten pathway: The complex cross talk between Notch and NF-κB. Lab. Investig. 2008, 88, 11–17. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Scott, O.; Roifman, C.M. NF-κB pathway and the Goldilocks principle: Lessons from human disorders of immunity and inflammation. J. Allergy Clin. Immunol. 2019, 143, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell. Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, X.; Ye, X.; Feng, C.; Yang, G.; Lu, Y.; Lin, Y.; Dong, C. High expression of stromal cell-derived factor 1 (SDF-1) and NF-κB predicts poor prognosis in cervical cancer. Med. Sci. Monit. 2017, 23, 151–157. [Google Scholar] [CrossRef][Green Version]
- Feng, W.; Xue, T.; Huang, S.; Shi, Q.; Tang, C.; Cui, G.; Yang, G.; Gong, H.; Guo, H. HIF-1α promotes the migration and invasion of hepatocellular carcinoma cells via the IL-8–NF-κB axis. Cell. Mol. Biol. Lett. 2018, 23, 26. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription factor C/EBP homologous protein in health and diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, N.; Ni, Y.S.; Yang, J.M.; Ma, L.; Lan, X.B.; Wu, J.; Niu, J.G.; Yu, J.Q. TRPM2 in ischemic stroke: Structure, molecular mechanisms, and drug intervention. Channels 2021, 15, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, J.; Jiang, N.; Xian, Y.; Ju, H.; Wei, Y.; Zhang, X. Corin protects H2O2-induced apoptosis through PI3K/AKT and NF-κB pathway in cardiomyocytes. Biomed. Pharmacother. 2018, 97, 594–599. [Google Scholar] [CrossRef]
- Qiu, X.; Liu, K.; Xiao, L.; Jin, S.; Dong, J.; Teng, X.; Guo, Q.; Chen, Y.; Wu, Y. Alpha-lipoic acid regulates the autophagy of vascular smooth muscle cells in diabetes by elevating hydrogen sulfide level. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3723–3738. [Google Scholar] [CrossRef]
- Wu, P.; Zeng, F.; Li, Y.X.; Yu, B.L.; Qiu, L.H.; Qin, W.; Li, J.; Zhou, Y.M.; Liang, F.R. Changes of resting cerebral activities in subacute ischemic stroke patients. Neural Regen. Res. 2015, 10, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, H.M.; Wilde, E.A.; Caeyenberghs, K.; Dennis, E.L. Longitudinal Neuroimaging in Pediatric Traumatic Brain Injury: Current State and Consideration of Factors That Influence Recovery. Front. Neurol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef]
- Clark, A.R.; Ohlmeyer, M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol. Ther. 2019, 201, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D.; Zachary, J.F. Nervous System. Pathol. Basis Vet. Dis. Expert Consult 2017, 14, 805–907.e1. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.P.; Henry, R.J.; Villapol, S.; Stoica, B.A.; Kumar, A.; Burns, M.P.; Faden, A.I.; Loane, D.J. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: Implications for traumatic brain injury. J. Neuroinflamm. 2017, 14, 65. [Google Scholar] [CrossRef]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.G.I.; Jain, P.; Khan, Z.K. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The role of astrocytes in neuroprotection after brain stroke: Potential in cell therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [PubMed]
- MUNTEANU, C.; TEOIBAS-SERBAN, D.; IORDACHE, L.; BALAUREA, M.; BLENDEA, C.-D. Water intake meets the Water from inside the human body—physiological, cultural, and health perspectives—Synthetic and Systematic literature review. Balneo PRM Res. J. 2021, 12, 196–209. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Gorgulho, C.M.; Romagnoli, G.G.; Bharthi, R.; Lotze, M.T. Johnny on the spot-chronic inflammation is driven by HMGB1. Front. Immunol. 2019, 10, 1561. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Choi, H.I.; Wang, Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H. A New Treatment Strategy for Parkinson’s Disease through the Gut–Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017, 26, 1560–1571. [Google Scholar] [CrossRef]
- Gou, X.; Ying, J.; Yue, Y.; Qiu, X.; Hu, P.; Qu, Y.; Li, J.; Mu, D. The Roles of High Mobility Group Box 1 in Cerebral Ischemic Injury. Front. Cell. Neurosci. 2020, 14, 280. [Google Scholar] [CrossRef]
- Ye, Y.; Zeng, Z.; Jin, T.; Zhang, H.; Xiong, X.; Gu, L. The role of high mobility group box 1 in ischemic stroke. Front. Cell. Neurosci. 2019, 13, 127. [Google Scholar] [CrossRef]
- Nekoui, A.; Blaise, G. Erythropoietin and Nonhematopoietic Effects. Am. J. Med. Sci. 2017, 353, 76–81. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Lukyanova, L.D.; Kirova, Y.I. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 320. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Olesch, C.; Kurrle, N.; Schnütgen, F.; Zukunft, S.; Fleming, I.; Brüne, B. Chronic Hypoxia Enhances β-Oxidation-Dependent Electron Transport via Electron Transferring Flavoproteins. Cells 2019, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ikeda, K.; Inoue, S. Functional mechanisms of mitochondrial respiratory chain supercomplex assembly factors and their involvement in muscle quality. Int. J. Mol. Sci. 2020, 21, 3182. [Google Scholar] [CrossRef] [PubMed]
- Brose, S.A.; Golovko, S.A.; Golovko, M.Y. Fatty acid biosynthesis inhibition increases reduction potential in neuronal cells under hypoxia. Front. Neurosci. 2016, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- Millar, L.J.; Shi, L.; Hoerder-Suabedissen, A.; Molnár, Z. Neonatal hypoxia ischaemia: Mechanisms, models, and therapeutic challenges. Front. Cell. Neurosci. 2017, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ferreira, E.; Hristova, M. Plasticity in the neonatal brain following hypoxic-ischaemic injury. Neural Plast. 2016, 2016, 4901014. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J. 2,4 Dinitrophenol as Medicine. Cells 2019, 8, 280. [Google Scholar] [CrossRef]
- Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive oxygen species-mediated damage of retinal neurons: Drug development targets for therapies of chronic neurodegeneration of the retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef]
- Weiskirchen, R. Hepatoprotective and anti-fibrotic agents: It’s time to take the next step. Front. Pharmacol. 2016, 6, 303. [Google Scholar] [CrossRef]
- Li, B.; Concepcion, K.; Meng, X.; Zhang, L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog. Neurobiol. 2017, 159, 50–68. [Google Scholar] [CrossRef]
- Bhalala, U.S.; Koehler, R.C.; Kannan, S. Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front. Pediatr. 2015, 2, 144. [Google Scholar] [CrossRef]
- de Faria, O.; Gonsalvez, D.G.; Nicholson, M.; Xiao, J. Activity-dependent central nervous system myelination throughout life. J. Neurochem. 2019, 148, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.; Wang, Y.; Akyol, O.; Ho, W.M.; Applegate, R.; Stier, G.; Martin, R.; Zhang, J.H. What’s new in traumatic brain injury: Update on tracking, monitoring and treatment. Int. J. Mol. Sci. 2015, 16, 11903–11965. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E. Cerebral blood flow and metabolism. Outcome Sev. Damage Cent. Nerv. Syst. 2008, 85–96. [Google Scholar] [CrossRef]
- Franke, H.; Illes, P. Nucleotide signaling in astrogliosis. Neurosci. Lett. 2014, 565, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Nas, K.; Yazmalar, L.; Şah, V.; Aydin, A.; Öneş, K. Rehabilitation of spinal cord injuries. World J. Orthop. 2015, 6, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, F.J.; Mattison, H.A.; Cerpa, W. Role of NMDA Receptor-Mediated Glutamatergic Signaling in Chronic and Acute Neuropathologies. Neural Plast. 2016, 2016, 2701526. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Pennisi, M.; Lanza, G.; Falzone, L.; Fisicaro, F.; Ferri, R.; Bella, R. Sars-cov-2 and the nervous system: From clinical features to molecular mechanisms. Int. J. Mol. Sci. 2020, 21, 5475. [Google Scholar] [CrossRef]
- Shahabipour, F.; Barati, N.; Johnston, T.P.; Derosa, G.; Maffioli, P.; Sahebkar, A. Exosomes: Nanoparticulate tools for RNA interference and drug delivery. J. Cell. Physiol. 2017, 232, 1660–1668. [Google Scholar] [CrossRef]
- Minhas, G.; Mathur, D.; Ragavendrasamy, B.; Sharma, N.K.; Paanu, V.; Anand, A. Hypoxia in CNS pathologies: Emerging role of miRNA-based Neurotherapeutics and yoga based alternative therapies. Front. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef] [PubMed]
- Morya, E.; Monte-Silva, K.; Bikson, M.; Esmaeilpour, Z.; Biazoli, C.E.; Fonseca, A.; Bocci, T.; Farzan, F.; Chatterjee, R.; Hausdorff, J.M.; et al. Beyond the target area: An integrative view of tDCS-induced motor cortex modulation in patients and athletes. J. Neuroeng. Rehabil. 2019, 16, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Li, L.; Li, X.; Wang, Q.; Ding, H.; Wang, X.; Ye, Z.; Wu, L.; Zhang, X.; et al. Sinomenine provides neuroprotection in model of traumatic brain injury via the Nrf2-ARE pathway. Front. Neurosci. 2016, 10, 580. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Andrews, A.M.; Paul, D.; Pachter, J.S. Extracellular vesicles: Mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 2018, 15, 19. [Google Scholar] [CrossRef]
- Iraci, N.; Leonardi, T.; Gessler, F.; Vega, B.; Pluchino, S. Focus on extracellular vesicles: Physiological role and signalling properties of extracellular membrane vesicles. Int. J. Mol. Sci. 2016, 17, 171. [Google Scholar] [CrossRef]
- Ciregia, F.; Urbani, A.; Palmisano, G. Extracellular vesicles in brain tumors and neurodegenerative diseases. Front. Mol. Neurosci. 2017, 10, 276. [Google Scholar] [CrossRef]
- Falkenberg, L.; Zeckey, C.; Mommsen, P.; Winkelmann, M.; Zelle, B.A.; Panzica, M.; Pape, H.C.; Krettek, C.; Probst, C. Long-term outcome in 324 polytrauma patients: What factors are associated with posttraumatic stress disorder and depressive disorder symptoms? Eur. J. Med. Res. 2017, 22, 44. [Google Scholar] [CrossRef]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Krawczyk, M.; Szukiewicz, D.; Gasik, R.; Kubaszewski, Ł.; Kurkowska-Jastrzębska, I. Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents. Mol. Neurobiol. 2017, 54, 2167–2188. [Google Scholar] [CrossRef]
- Lin, W.; Stone, S. Unfolded protein response in myelin disorders. Neural Regen. Res. 2020, 15, 636–645. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, J.S.; An, S.S.; Pennant, W.A.; Gwak, S.J.; Kim, A.N.; Han, P.K.; Yoon, D.H.; Kim, K.N.; Ha, Y. Hypoxia-specific GM-CSF-overexpressing neural stem cells improve graft survival and functional recovery in spinal cord injury. Gene Ther. 2012, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Che, L.; Lee, H.Y.; Lee, H.L.; Yun, Y.; Lee, M.; Oh, J.; Ha, Y. Antiapoptotic effect of highly secreted GMCSF from neuronal cell-specific GMCSF overexpressing neural stem cells in spinal cord injury model. Spine 2015, 40, E1284–E1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, J.Y.; Kim, J.H.; Jung, H.; Lee, W.T.; Lee, J.E. Restorative mechanism of neural progenitor cells overexpressing arginine decarboxylase genes following ischemic injury. Exp. Neurobiol. 2019, 28, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lou, X.; Xu, S.; Du, J.; Wu, J. Hypoxia inducible factor-1 (HIF-1α) reduced inflammation in spinal cord injury via miR-380-3p/ NLRP3 by Circ 0001723. Biol. Res. 2020, 53, 1–14. [Google Scholar] [CrossRef]
- Zhong, D.; Cao, Y.; Li, C.J.; Li, M.; Rong, Z.J.; Jiang, L.; Guo, Z.; Lu, H.B.; Hu, J.Z. Highlight article: Neural stem cell-derived exosomes facilitate spinal cord functional recovery after injury by promoting angiogenesis. Exp. Biol. Med. 2020, 245, 54–65. [Google Scholar] [CrossRef]
- Long, H.Q.; Li, G.S.; Cheng, X.; Xu, J.H.; Li, F.B. Role of hypoxia-induced VEGF in blood-spinal cord barrier disruption in chronic spinal cord injury. Chin. J. Traumatol. Engl. Ed. 2015, 18, 293–295. [Google Scholar] [CrossRef]
- Li, Y.; Han, W.; Wu, Y.; Zhou, K.; Zheng, Z.; Wang, H.; Xie, L.; Li, R.; Xu, K.; Liu, Y.; et al. Stabilization of Hypoxia Inducible Factor-1α by Dimethyloxalylglycine Promotes Recovery from Acute Spinal Cord Injury by Inhibiting Neural Apoptosis and Enhancing Axon Regeneration. J. Neurotrauma 2019, 36, 3394–3409. [Google Scholar] [CrossRef]
- Wang, X.; Ma, J.; Fu, Q.; Zhu, L.; Zhang, Z.; Zhang, F.; Lu, N.; Chen, A. Role of hypoxia-inducible factor-1α in autophagic cell death in microglial cells induced by hypoxia. Mol. Med. Rep. 2017, 15, 2097–2105. [Google Scholar] [CrossRef]
- Dale-Nagle, E.A.; Hoffman, M.S.; MacFarlane, P.M.; Satriotomo, I.; Lovett-Barr, M.R.; Vinit, S.; Mitchell, G.S. Spinal plasticity following intermittent hypoxia: Implications for spinal injury. Ann. N. Y. Acad. Sci. 2010, 1198, 252–259. [Google Scholar] [CrossRef]
- Perim, R.R.; Mitchell, G.S. Circulatory control of phrenic motor plasticity. Respir. Physiol. Neurobiol. 2019, 265, 19–23. [Google Scholar] [CrossRef]
- Wen, M.H.; Wu, M.J.; Vinit, S.; Lee, K.Z. Modulation of Serotonin and Adenosine 2A Receptors on Intermittent Hypoxia-Induced Respiratory Recovery following Mid-Cervical Contusion in the Rat. J. Neurotrauma 2019, 36, 2991–3004. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Astorino, T.A.; Harness, E.T.; White, A.C. Efficacy of acute intermittent hypoxia on physical function and health status in humans with spinal cord injury: A brief review. Neural Plast. 2015, 2015, 409625. [Google Scholar] [CrossRef] [PubMed]
- Naidu, A.; Peters, D.M.; Tan, A.Q.; Barth, S.; Crane, A.; Link, A.; Balakrishnan, S.; Hayes, H.B.; Slocum, C.; Zafonte, R.D.; et al. Daily acute intermittent hypoxia to improve walking function in persons with subacute spinal cord injury: A randomized clinical trial study protocol. BMC Neurol. 2020, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rothi, E.J.; Lee, K.Z.; Dale, E.A.; Reier, P.J.; Mitchell, G.S.; Fuller, D.D. Intermittent hypoxia and neurorehabilitation. J. Appl. Physiol. 2015, 119, 1455–1465. [Google Scholar] [CrossRef]
- Vinit, S.; Lovett-Barr, M.R.; Mitchell, G.S. Intermittent hypoxia induces functional recovery following cervical spinal injury. Respir. Physiol. Neurobiol. 2009, 169, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, B.J.; Terada, J.; Springborn, S.R.; Vinit, S.; MacFarlane, P.M.; Mitchell, G.S. Daily acute intermittent hypoxia improves breathing function with acute and chronic spinal injury via distinct mechanisms. Respir. Physiol. Neurobiol. 2018, 256, 50–57. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, P.; Peng, Y.; Ouyang, R. Role of Oxidative Stress in the Neurocognitive Dysfunction of Obstructive Sleep Apnea Syndrome. Oxidative Med. Cell. Longev. 2016, 2016, 9626831. [Google Scholar] [CrossRef]
- Vivodtzev, I.; Tan, A.Q.; Hermann, M.; Jayaraman, A.; Stahl, V.; Rymer, W.Z.E.V.; Mitchell, G.S.; Hayes, H.B.; Trumbower, R.D. Mild to moderate sleep apnea is linked to hypoxia-induced motor recovery after spinal cord injury. Am. J. Respir. Crit. Care Med. 2020, 202, 887–890. [Google Scholar] [CrossRef]
- Ke, P.Y. Diverse functions of autophagy in liver physiology and liver diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef]
- Xiong, T.; Yang, X.; Qu, Y.; Chen, H.; Yue, Y.; Wang, H.; Zhao, F.; Li, S.; Zou, R.; Zhang, L.; et al. Erythropoietin induces synaptogenesis and neurite repair after hypoxia ischemia-mediated brain injury in neonatal rats. Neuroreport 2019, 30, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Kant, R.; Milner, R. Chronic mild hypoxia promotes profound vascular remodeling in spinal cord blood vessels, preferentially in white matter, via an α5β1 integrin-mediated mechanism. Angiogenesis 2018, 21, 251–266. [Google Scholar] [CrossRef]
- Munteanu, C. Cell biology considerations in Spinal Cord Injury—Review. Balneo Res. J. 2017, 8, 136–151. [Google Scholar] [CrossRef]
- Tang, G.; Chen, Y.; Chen, J.; Chen, Z.; Jiang, W. Deferoxamine Ameliorates Compressed Spinal Cord Injury by Promoting Neovascularization in Rats. J. Mol. Neurosci. 2020, 70, 1437–1444. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Green, P.; Tapoulal, N.; Lewandowski, A.J.; Leeson, P.; Herring, N. The role of neuropeptide Y in cardiovascular health and disease. Front. Physiol. 2018, 9, 1281. [Google Scholar] [CrossRef] [PubMed]
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of hydrogen sulfide against the progression of severe Alzheimer’s disease in transgenic mice at different ages. Pharmacology 2019, 103, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Yung Justin Hou Ming, G.A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, X.; Huang, D.; Luo, Q.; Zheng, M.; Wang, K.; Cao, L.; Yin, Z. Erythropoietin signaling increases neurogenesis and oligodendrogenesis of endogenous neural stem cells following spinal cord injury both in vivo and in vitro. Mol. Med. Rep. 2018, 17, 264–272. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Matsumoto, M.; Ichikawa, T.; Young, W.; Kodama, N. Glutamine synthetase protects the spinal cord against hypoxia-induced and GABAA receptor-activated axonal depressions. Surg. Neurol. 2008, 70, 122–128. [Google Scholar] [CrossRef]
- Gaforio, J.J.; Visioli, F.; Alarcón-De-la-lastra, C.; Castañer, O.; Delgado-Rodríguez, M.; Fitó, M.; Hernández, A.F.; Huertas, J.R.; Martínez-González, M.A.; Menendez, J.A.; et al. Virgin olive oil and health: Summary of the iii international conference on virgin olive oil and health consensus report, JAEN (Spain) 2018. Nutrients 2019, 11, 2039. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Barbalace, M.C.; Hrelia, S. Bioactivity of olive oil phenols in neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef]
- Libro, R.; Giacoppo, S.; Rajan, T.S.; Bramanti, P.; Mazzon, E. Natural phytochemicals in the treatment and prevention of dementia: An overview. Molecules 2016, 21, 518. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, D.M. Antioxidant therapies for neuroprotection—A review. J. Clin. Med. 2019, 8, 1659. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 733. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Molina-Holgado, E.; Molina-Holgado, F. Effect of endocannabinoid signalling on cell fate: Life, death, differentiation and proliferation of brain cells. Br. J. Pharmacol. 2019, 176, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Guzman, M.; Mackie, K.; Doherty, P.; Sciences, B.; Kingdom, U.; Institutet, K.; Maccarrone, M.; Guzmán, M.; Mackie, K.; et al. Programming of neural cells by (endo)cannabinoids: From physiological rules to emerging therapies. Nat. Rev. Neurosci. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Han, S.M.; Park, K.K. Therapeutic effects of apamin as a bee venom component for non-neoplastic disease. Toxins 2020, 12, 195. [Google Scholar] [CrossRef]
- Cramer, S.W.; Chen, C.C. Photodynamic Therapy for the Treatment of Glioblastoma. Front. Surg. 2020, 6, 81. [Google Scholar] [CrossRef]
- Munteanu, C.; Munteanu, D.; Onose, G. Hydrogen sulfide (H2S)—Therapeutic relevance in rehabilitation and balneotherapy Systematic literature review and meta-analysis based on the PRISMA paradig. Balneo PRM Res. J. 2021, 12, 176–195. [Google Scholar] [CrossRef]
- Firan, F.C.; Romila, A.; Onose, G. Current synthesis and systematic review of main effects of calf blood deproteinized medicine (Actovegin® ) in ischemic stroke. Int. J. Mol. Sci. 2020, 21, 3181. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Yao, G.; Liu, Z.; Cui, R.; Yang, W. Mechanisms of Transcranial Magnetic Stimulation Treating on Post-stroke Depression. Front. Hum. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Dogaru, G.; Rotariu, M.; Onose, G. Therapeutic gases used in balneotherapy and rehabilitation medicine—Scientific relevance in the last ten years (2011–2020)—Synthetic literature review. Balneo PRM Res. J. 2021, 12, 111–122. [Google Scholar] [CrossRef]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef]
- Murugesan, T.; Rajajeyabalachandran, G.; Kumar, S.; Nagaraju, S.; Jegatheesan, S.K. Targeting HIF-2α as therapy for advanced cancers. Drug Discov. Today 2018, 23, 1444–1451. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoica, S.I.; Bleotu, C.; Ciobanu, V.; Ionescu, A.M.; Albadi, I.; Onose, G.; Munteanu, C. Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery. Biomedicines 2022, 10, 481. https://doi.org/10.3390/biomedicines10020481
Stoica SI, Bleotu C, Ciobanu V, Ionescu AM, Albadi I, Onose G, Munteanu C. Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery. Biomedicines. 2022; 10(2):481. https://doi.org/10.3390/biomedicines10020481
Chicago/Turabian StyleStoica, Simona Isabelle, Coralia Bleotu, Vlad Ciobanu, Anca Mirela Ionescu, Irina Albadi, Gelu Onose, and Constantin Munteanu. 2022. "Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery" Biomedicines 10, no. 2: 481. https://doi.org/10.3390/biomedicines10020481
APA StyleStoica, S. I., Bleotu, C., Ciobanu, V., Ionescu, A. M., Albadi, I., Onose, G., & Munteanu, C. (2022). Considerations about Hypoxic Changes in Neuraxis Tissue Injuries and Recovery. Biomedicines, 10(2), 481. https://doi.org/10.3390/biomedicines10020481