
Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Laboratory Measurements
2.3. Statistical Analysis
3. Results
3.1. Baseline Characteristics of Patients Studied
3.2. Correlations between Metabolic Parameters at Baseline
3.3. Changes in Metabolic Parameters after the Start of Pemafibrate
3.3.1. Changes in Metabolic Parameters after the Start of Pemafibrate in All Patients
3.3.2. Changes in Metabolic Parameters after the Start of Pemafibrate in Patients with and without the Treatment Using Sodium-Glucose Co-Transporter 2 Inhibitors (SGLT2i)
3.4. Correlations between Changes in Metabolic Parameters after the Start of Pemafibrate
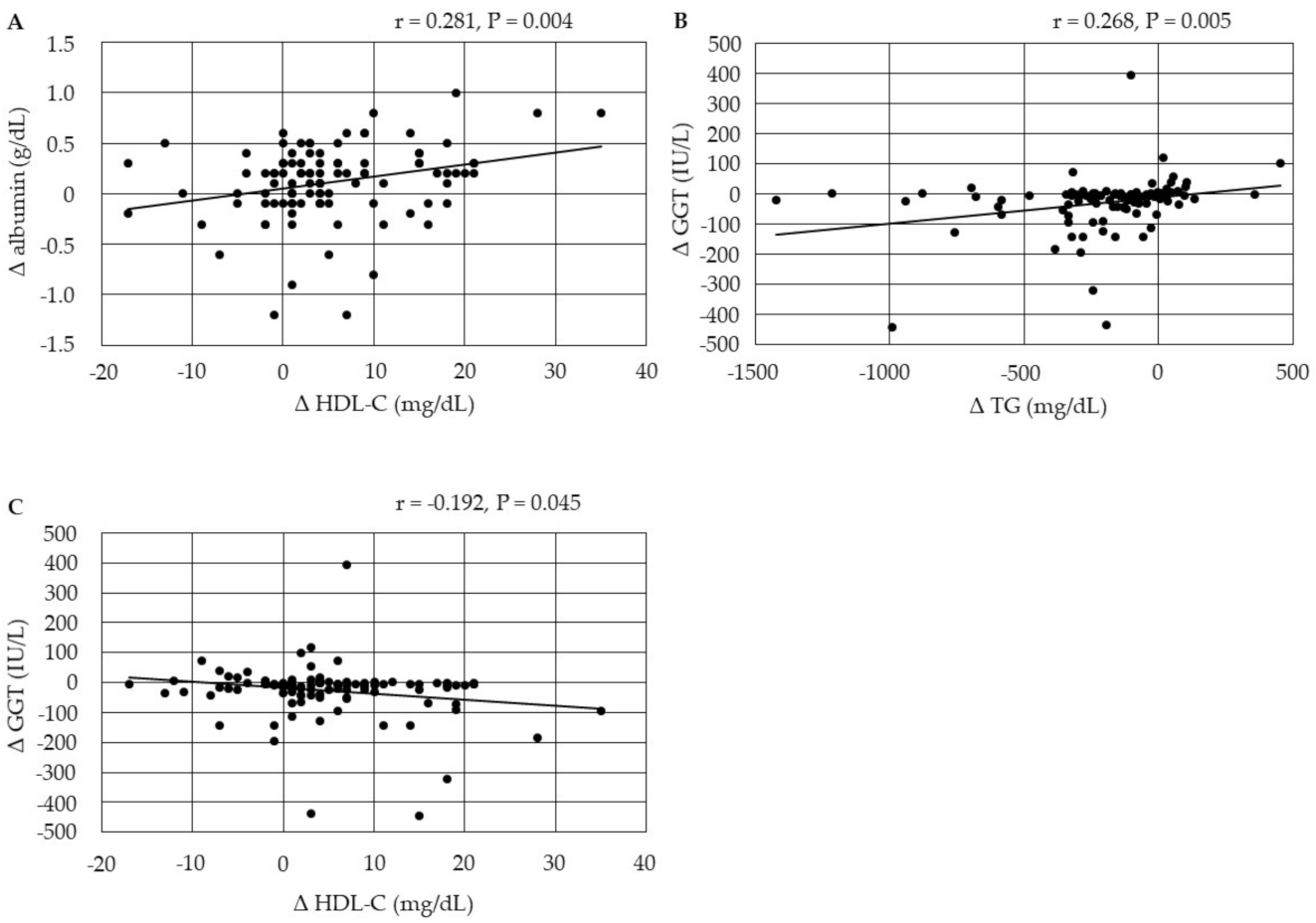
3.4.1. Correlations between Changes in Serum Lipids
3.4.2. Correlations among Changes in the Markers for Liver Function
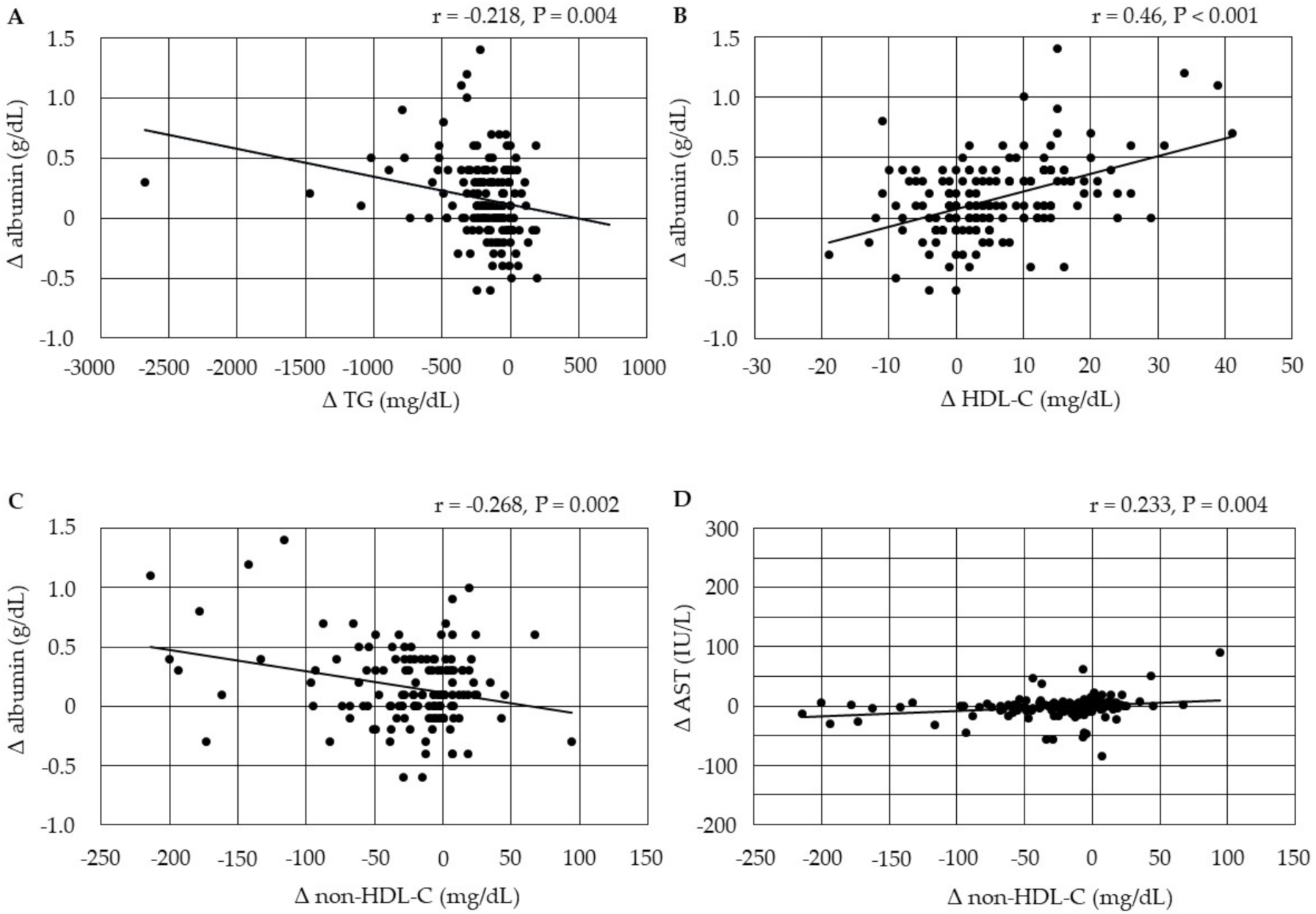
3.4.3. Correlations between Changes in Serum Lipids and Changes in the Markers for Liver Function
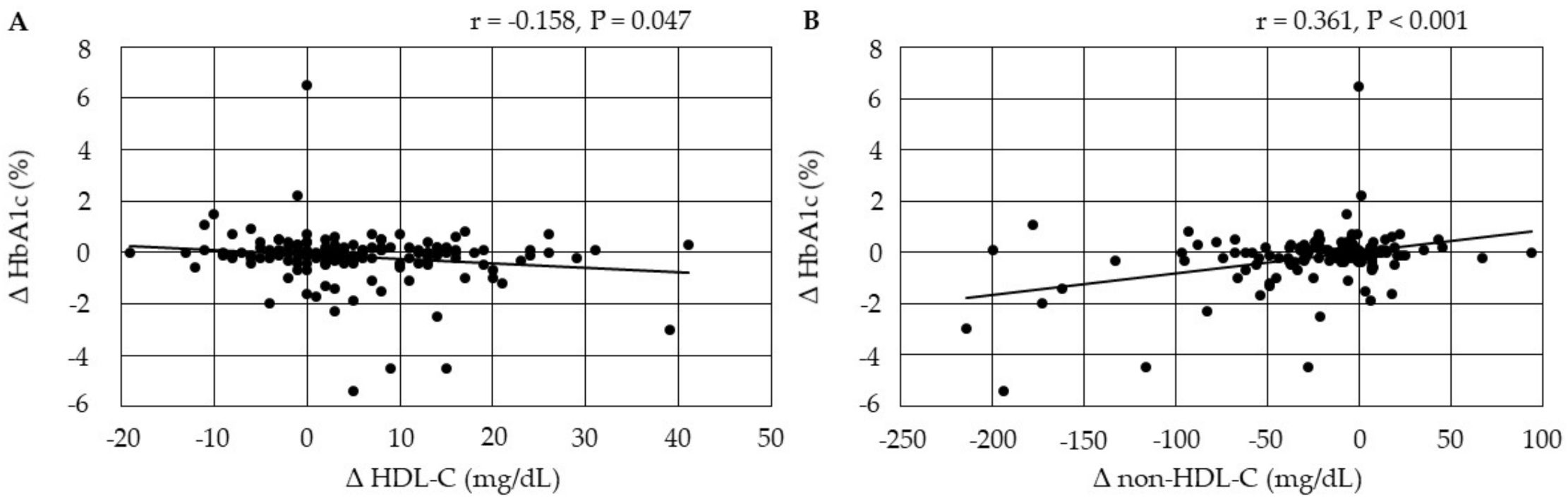
3.4.4. Correlations between Changes in Serum Lipids and Changes in HbA1c after the Start of Pemafibrate
3.4.5. Correlations between Changes in Serum Lipids and Changes in Serum UA after the Start of Pemafibrate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed]
- Motojima, K. Peroxisome Proliferator-Activated Receptor (PPAR): Structure, Mechanisms of Activation and Diverse Functions. Cell Struct. Funct. 1993, 18, 267–277. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wójtowicz, S.; Strosznajder, J.B.; Jeżyna, M. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C. Selective peroxisome proliferator-activated receptor α modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc. Diabetol. 2013, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Daua, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.S.; Arora, R. Prevention of Cardiovascular Disease Utilizing Fibrates-A Pooled Meta-analysis. Am. J. Ther. 2010, 17, e182–e188. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S. Fenofibrate: Treatment of hyperlipidemia and beyond. Expert Rev. Cardiovasc. Ther. 2008, 6, 1319–1330. [Google Scholar] [CrossRef]
- Feingold, K.; Grunfeld, C. Triglyceride Lowering Drugs. In Endotext; Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2017. [Google Scholar]
- Wang, D.; Liu, B.; Tao, W.; Hao, Z.; Liu, M. Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst. Rev. 2015, 2018, CD009580. [Google Scholar] [CrossRef]
- Ahmad, J.; Odin, J.A.; Hayashi, P.H.; Chalasani, N.; Fontana, R.J.; Barnhart, H.; Cirulli, E.T.; Kleiner, D.E.; Hoofnagle, J.H. Identification and Characterization of Fenofibrate-Induced Liver Injury. Am. J. Dig. Dis. 2017, 62, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; Armani, A.; McKenney, J.M.; Jacobson, T. Safety Considerations with Fibrate Therapy. Am. J. Cardiol. 2007, 99, S3–S18. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor α agonists. Bioorg. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S.; on behalf of the KSGOB. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-alpha modulator (SPPARMalpha), in dyslipidemic patients with renal impairment. Int. J. Mol. Sci. 2019, 20, 706. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Clinical applications of a novel selective PPARalpha modulator, pemafibrate, in dyslipidemia and metabolic diseases. J. Atheroscler. Thromb. 2019, 26, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Kawaguchi, A.; Sako, A.; Mishima, S.; Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters in Patients with Type 2 Diabetes: A Chart-Based Analysis. J. Clin. Med. Res. 2016, 8, 237–243. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Kawaguchi, A.; Waragai, Y.; Harigae, T.; Masui, Y.; Kakuta, K.; Hamasaki, H.; Katsuyama, H.; et al. Effects of Six Kinds of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters, and Summarized Effect and Its Correlations with Baseline Data. J. Clin. Med. Res. 2017, 9, 605–612. [Google Scholar] [CrossRef][Green Version]
- Katsuyama, H.; Hakoshima, M.; Iijima, T.; Adachi, H.; Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Hepatic Fibrosis in Patients with Type 2 Diabetes: A Chart-Based Analysis. J. Endocrinol. Metab. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Fan, J.-G.; Peng, Y.-D. Metabolic syndrome and non-alcoholic fatty liver disease: Asian definitions and Asian studies. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 572–578. [Google Scholar]
- Yanai, H.; Hirowatari, Y.; Yoshida, H. Diabetic dyslipidemia: Evaluation and mechanism. Glob. Health Med. 2019, 1, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hirowatari, Y.; Ito, K.; Kurosawa, H.; Tada, N.; Yoshida, H. Understanding of Diabetic Dyslipidemia by Using the Anion-Exchange High Performance Liquid Chromatography Data. J. Clin. Med. Res. 2016, 8, 424–426. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sztalryd, C.; Kraemer, F. Regulation of hormone-sensitive lipase in streptozotocin-induced diabetic rats. Metabolism 1995, 44, 1391–1396. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular apoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2001, 276, 20803–20804. [Google Scholar] [CrossRef]
- Chen, M.; Breslow, J.L.; Li, W.; Leff, T. Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: Correlation with changes in plasma triglyceride levels. J. Lipid Res. 1994, 35, 1918–1924. [Google Scholar] [CrossRef]
- Fisher, E.A. The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 778–781. [Google Scholar] [CrossRef]
- Nikkilä, E.A.; Taskinen, M.-R.; Kekki, M. Relation of plasma high-density lipoprotein cholesterol to lipoprotein-lipase activity in adipose tissue and skeletal muscle of man. Atherosclerosis 1978, 29, 497–501. [Google Scholar] [CrossRef]
- Ooi, E.M.M.; Barrett, P.H.R.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an emerging cardiovascular risk factor. Clin. Sci. 2008, 114, 611–624. [Google Scholar] [CrossRef]
- Engin, A. Non-Alcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2017, 960, 443–467. [Google Scholar]
- Paiva, A.A.; Raposo, H.F.; Wanschel, A.C.; Nardelli, T.R.; Oliveira, H.C. Apolipoprotein CIII Overexpression-Induced Hypertriglyceridemia Increases Nonalcoholic Fatty Liver Disease in Association with Inflammation and Cell Death. Oxid. Med. Cell. Longev. 2017, 2017, 1838679. [Google Scholar] [CrossRef]
- Giby, V.G.; Ajith, T.A. Role of adipokines and peroxisome proliferator-activated receptors in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 570–579. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yu, C.; Li, X.; Sun, L.; Zhu, X.; Zhao, C.; Zhang, Z.; Yang, Z. Serum Uric Acid Levels and Risk of Metabolic Syndrome: A Dose-Response Meta-Analysis of Prospective Studies. J. Clin. Endocrinol. Metab. 2015, 100, 4198–4207. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate–anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sanna, S.; Maschio, A.; Busonero, F.; Usala, G.; Mulas, A.; Lai, S.; Dei, M.; Orrù, M.; Albai, G. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007, 3, e194. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.; Floyd, J.; Palmer, C.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Doshi, M.; Takiue, Y.; Saito, H.; Hosoyamada, M. The Increased Protein Level of URAT1 was Observed in Obesity/Metabolic Syndrome Model Mice. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1290–1294. [Google Scholar] [CrossRef]
- Miao, Z.; Yan, S.; Wang, J.; Wang, B.; Li, Y.; Xing, X.; Yuan, Y.; Meng, D.; Wang, L.; Gu, J. Insulin resistance acts as an independent risk factor exacerbating high-purine diet induced renal injury and knee joint gouty lesions. Inflamm. Res. 2009, 58, 659–668. [Google Scholar] [CrossRef]
- Ng, H.-Y.; Lee, Y.-T.; Kuo, W.-H.; Huang, P.-C.; Lee, W.-C.; Lee, C.-T. Alterations of Renal Epithelial Glucose and Uric Acid Transporters in Fructose Induced Metabolic Syndrome. Kidney Blood Press. Res. 2018, 43, 1822–1831. [Google Scholar] [CrossRef]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef] [PubMed]
- Leyva, F.; Wingrove, C.S.; Godsland, I.F.; Stevenson, J.C. The glycolytic pathway to coronary heart disease: A hypothesis. Metabolism 1998, 47, 657–662. [Google Scholar] [CrossRef]
- Neve, B.; Fruchart, J.-C.; Staels, B. Role of the peroxisome proliferator-activated receptors (PPAR) in atherosclerosis. Biochem. Pharmacol. 2000, 60, 1245–1250. [Google Scholar] [CrossRef]
- Ge, P.-L.; Du, S.-D.; Mao, Y.-L. Advances in preoperative assessment of liver function. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 361–370. [Google Scholar] [CrossRef]
- Guo, R.; Liong, E.C.; So, K.F.; Fung, M.-L.; Tipoe, G.L. Beneficial mechanisms of aerobic exercise on hepatic lipid metabolism in non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 139–144. [Google Scholar] [CrossRef]
- Kim, H.; Haluzik, M.; Asghar, Z.; Yau, D.; Joseph, J.W.; Fernandez, A.M.; Reitman, M.L.; Yakar, S.; Stannard, B.; Heron-Milhavet, L.; et al. Peroxisome Proliferator–Activated Receptor-α Agonist Treatment in a Transgenic Model of Type 2 Diabetes Reverses the Lipotoxic State and Improves Glucose Homeostasis. Diabetes 2003, 52, 1770–1778. [Google Scholar] [CrossRef]
- Leclercq, I.A. Pathogenesis of steatohepatitis: Insights from the study of animal models. Acta Gastro Enterol. Belg. 2007, 70, 25–31. [Google Scholar]
- Ahima, R.S. Adipose Tissue as an Endocrine Organ. Obesity 2006, 14, 242S–249S. [Google Scholar] [CrossRef] [PubMed]
- Giralt, M.; Villarroya, F. White, Brown, Beige/Brite: Different Adipose Cells for Different Functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. 2007, 83 (Suppl. 5), S192–S203. [Google Scholar] [CrossRef]
- Kim, S.H.; Jorge Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-alpha and PPAR-beta/delta on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365. [Google Scholar] [CrossRef] [PubMed]
- Maia-Fernandes, T.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Cardiovascular actions of adiponectin: Pathophysiologic implications. Rev. Port. Cardiol. 2008, 27, 1431–1449. [Google Scholar] [PubMed]
- Yanai, H.; Yoshida, H. Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef]
- Haluzík, M.M.; Haluzík, M. PPAR-alpha and insulin sensitivity. Physiol. Res. 2005, 55, 115–122. [Google Scholar]
- Uetake, D.; Ohno, I.; Ichida, K.; Yamaguchi, Y.; Saikawa, H.; Endou, H.; Hosoya, T. Effect of fenofibrate on uric acid metabolism and urate transporter 1. Intern. Med. 2010, 49, 89–94. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, X.; Dong, Z.; Lu, J.; Zhao, Y.; Li, R.; Li, C.; Chen, Y. Impact of fenofibrate therapy on serum uric acid concentrations: A review and meta-analysis. Endocr. J. 2021, 68, 829–837. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Plasma uric acid concentrations are reduced by fenofibrate: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol. Res. 2015, 102, 63–70. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H.; Hakoshima, M. A Significant Increase of Estimated Glomerular Filtration Rate After Switching From Fenofibrate to Pemafibrate in Type 2 Diabetic Patients. Cardiol. Res. 2021, 12, 358–362. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | |
|---|---|
| Age (years old) | 60.0 ± 15.9 |
| Gender (male/female) | 140/106 |
| Body weight (mean ± SD, kg) | 73.7 ± 16.6 |
| Body mass index (mean ± SD, kg/m2) | 27.0 ± 5.5 |
| Systolic blood pressure (mean ± SD, mmHg) | 134.1 ± 17.3 |
| Diastolic blood pressure (mean ± SD, mmHg) | 78.4 ± 13.9 |
| Comorbidities | |
| Type 2 diabetes (n, %) | 121, 49.2% |
| Hypertension (n, %) | 113, 46.0% |
| Hyperuricemia (n, %) | 73, 29.7% |
| Treatments for type 2 diabetes | |
| Dipeptidyl peptidase-4 inhibitors (n, %) | 67, 27.2% |
| Metformin (n, %) | 76, 30.9% |
| Sodium-glucose co-transporter 2 inhibitors (n, %) | 63, 25.6% |
| Pioglitazone (n, %) | 18, 7.3% |
| Insulin (n, %) | 23, 9.3% |
| Glucagon-like peptide 1 receptor-agonists (n, %) | 17, 6.9% |
| Treatments for hypertension | |
| Angiotensin receptor blockers (n, %) | 88, 35.8% |
| Calcium antagonists (n, %) | 95, 38.6% |
| Diuretics (n, %) | 14, 5.7% |
| α, β-blockers (n, %) | 31, 12.6% |
| Treatments for dyslipidemia | |
| Statins (n, %) | 85, 34.6% |
| Ezetimibe (n) | 43, 17.5% |
| Eicosapentaenoic acid (n, %) | 30, 12.2% |
| Fenofibrate (n, %) | 20, 8.1% |
| Treatments for hyperuricemia | |
| Febuxostat (n, %) | 22, 8.9% |
| Topiroxostat (n, %) | 8, 3.3% |
| Allopurinol (n, %) | 12, 4.9% |
| Baseline | After 3 Months | After 6 Months | After 12 Months | |
|---|---|---|---|---|
| Body weight (kg) | 73.7 ± 16.7 | 72.9 ± 15.2 | 73.1 ± 14.0 | 73.3 ± 14.4 |
| Systolic blood pressure (mmHg) | 134.1 ± 17.3 | 133.0 ± 14.5 | 133.9 ± 14.2 | 132.0 ± 14.2 |
| Diastolic blood pressure (mmHg) | 78.4 ± 13.9 | 78.6 ± 12.0 | 78.7 ± 12.0 | 77.6 ± 12.0 |
| TG (mg/dL) | 411.0 ± 349.5 | 231.1 ± 221.9 * | 214.2 ± 171.6 * | 206.1 ± 147.8 * |
| HDL-C (mg/dL) | 46.0 ± 13.7 | 51.0 ± 13.3 * | 50.5 ± 12.7 * | 51.4 ± 13.2 * |
| LDL-C (mg/dL) | 105.6 ± 35.3 | 110.7 ± 33.1 | 107.0 ± 31.6 | 109.1 ± 32.6 |
| Non-HDL-C (mg/dL) | 170.6 ± 56.3 | 147.3 ± 39.5 * | 141.2 ± 36.8 * | 138.6 ± 35.5 * |
| AST (IU/L) | 36.5 ± 58.1 | 33.9 ± 48.4 * | 29.7 ± 20.2 * | 28.9 ± 21.2 |
| ALT (IU/L) | 40.6 ± 42.3 | 33.5 ± 32.4 * | 30.0 ± 24.5 * | 30.5 ± 31.3 * |
| GGT (IU/L) | 80.4 ± 129.3 | 50.5 ± 70.7 * | 57.8 ± 87.7 * | 53.0 ± 74.6 * |
| Albumin (g/dL) | 4.25 ± 0.44 | 4.43 ± 0.36 * | 4.42 ± 0.37 * | 4.42 ± 0.38 * |
| UA (mg/dL) | 6.0 ± 1.6 | 6.0 ± 1.5 | 5.8 ± 1.5 | 5.7 ± 1.2 * |
| eGFR (mL/min/1.73m2) | 72.6 ± 27.7 | 72.1 ± 28.6 | 71.5 ± 23.7 | 68.1 ± 21.9 |
| Plasma glucose (mg/dL) | 142.4 ± 49.5 | 137.6 ± 43.8 | 138.3 ± 48.4 | 135.0 ± 38.2 |
| HbA1c (%) | 6.8 ± 1.3 | 6.6 ± 1.0 * | 6.6 ± 0.9 | 6.6 ± 0.9 |
| Patients without the Treatment Using SGLT2i (n = 183) | Patients with the Treatment Using SGLT2i (n = 63) | |||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | After 3 Months | After 6 Months | After 12 Months | Baseline | After 3 Months | After 6 Months | After 12 Months | |
| Body weight (kg) | 69.4 ± 17.6 | 71.6 ± 15.8 | 72.3 ± 14.9 | 73.2 ± 15.4 | 77.3 ± 14.2 | 76.2 ± 13.3 | 75.0 ± 12.1 | 73.1 ± 11.6 |
| Systolic blood pressure (mmHg) | 133.4 ± 18.3 | 132.2 ± 14.0 | 133.5 ± 14.2 | 132.7 ± 14.9 | 133.1 ± 17.9 | 133.5 ± 15.8 | 133.8 ± 14.2 | 130.7 ± 11.5 |
| Diastolic blood pressure (mmHg) | 77.4 ± 12.9 | 77.4 ± 12.4 | 77.4 ± 11.8 | 77.2 ± 12.7 | 81.2 ± 14.4 | 81.0 ± 10.9 | 80.9 ± 12.1 | 79.4 ± 10.5 |
| TG (mg/dL) | 376.6 ± 342.3 | 208.0 ± 167.1 * | 202.3 ± 139.6 * | 196.3 ± 143.1 * | 422.5 ± 327.4 | 298.0 ± 323.7 * | 259.7 ± 248.2 * | 235.4 ± 158.8 * |
| HDL-C (mg/dL) | 47.0 ± 15.1 | 51.9 ± 13.5 * | 51.1 ± 13.5 * | 51.8 ± 13.2 * | 46.7 ± 12.2 | 49.3 ± 13.0 * | 49.4 ± 11.3 * | 51.7 ± 11.3 * |
| LDL-C (mg/dL) | 107.1 ± 33.5 | 114.0 ± 34.8 * | 111.4 ± 34.8 | 109.3 ± 34.2 * | 105.7 ± 35.3 | 104.3 ± 29.3 | 99.0 ± 28.9 | 104.4 ± 29.5 |
| Non-HDL-C (mg/dL) | 166.5 ± 50.5 | 148.1 ± 39.6 * | 143.4 ± 37.9 * | 138.1 ± 38.1 * | 167.4 ± 60.6 | 143.0 ± 39.1 * | 130.5 ± 31.0 * | 133.2 ± 30.1 * |
| AST (IU/L) | 35.9 ± 60.9 | 35.9 ± 53.9 | 31.3 ± 23.0 | 31.3 ± 29.8 | 35.0 ± 26.6 | 28.8 ± 25.2 * | 26.9 ± 13.4 | 27.8 ± 15.3 * |
| ALT (IU/L) | 38.1 ± 41.5 | 33.4 ± 28.3 * | 30.7 ± 25.8 * | 30.3 ± 32.1 * | 41.4 ± 34.5 | 34.2 ± 41.9 * | 29.1 ± 19.8 * | 32.4 ± 30.1 * |
| GGT (IU/L) | 80.3 ± 143.6 | 52.8 ± 78.3 * | 61.4 ± 106.3 * | 55.7 ± 135.2 * | 88.3 ± 118.8 | 43.6 ± 38.9 * | 55.9 ± 66.9 * | 78.4 ± 110.9 * |
| Albumin (g/dL) | 4.22 ± 0.42 | 4.43 ± 0.36 * | 4.41 ± 0.37 * | 4.44 ± 0.33 * | 4.33 ± 0.45 | 4.43 ± 0.36 * | 4.47 ± 0.37 | 4.40 ± 0.47 |
| UA (mg/dL) | 6.0 ± 1.7 | 6.1 ± 1.5 | 5.9 ± 1.6 | 5.8 ± 1.3 * | 5.8 ± 1.5 | 5.9 ± 1.6 | 5.5 ± 1.2 | 5.5 ± 1.0 |
| eGFR (mL/min/1.73m2) | 69.8 ± 24.7 | 69.5 ± 18.6 | 70.1 ± 19.3 | 66.2 ± 18.6 | 78.5 ± 34.3 | 81.9 ± 44.1 | 79.2 ± 33.7 | 75.7 ± 29.5 |
| Plasma glucose (mg/dL) | 134.7 ± 51.3 | 130.1 ± 42.3 | 132.4 ± 47.9 | 130.4 ± 38.7 | 163.3 ± 49.7 | 153.2 ± 42.9 * | 151.7 ± 46.1 | 146.2 ± 34.2 |
| HbA1c (%) | 6.3 ± 1.1 | 6.3 ± 0.8 | 6.3 ± 0.8 | 6.3 ± 0.8 | 7.6 ± 1.3 | 7.2 ± 1.0 * | 7.2 ± 0.9 | 7.2 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Katsuyama, H.; Hakoshima, M. Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines 2022, 10, 401. https://doi.org/10.3390/biomedicines10020401
Yanai H, Katsuyama H, Hakoshima M. Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines. 2022; 10(2):401. https://doi.org/10.3390/biomedicines10020401
Chicago/Turabian StyleYanai, Hidekatsu, Hisayuki Katsuyama, and Mariko Hakoshima. 2022. "Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study" Biomedicines 10, no. 2: 401. https://doi.org/10.3390/biomedicines10020401
APA StyleYanai, H., Katsuyama, H., & Hakoshima, M. (2022). Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines, 10(2), 401. https://doi.org/10.3390/biomedicines10020401