
Identification of Potential Targets Linked to the Cardiovascular/Alzheimer’s Axis through Bioinformatics Approaches

 , ,
, ,  , , and
, , and 
Abstract
1. Introduction
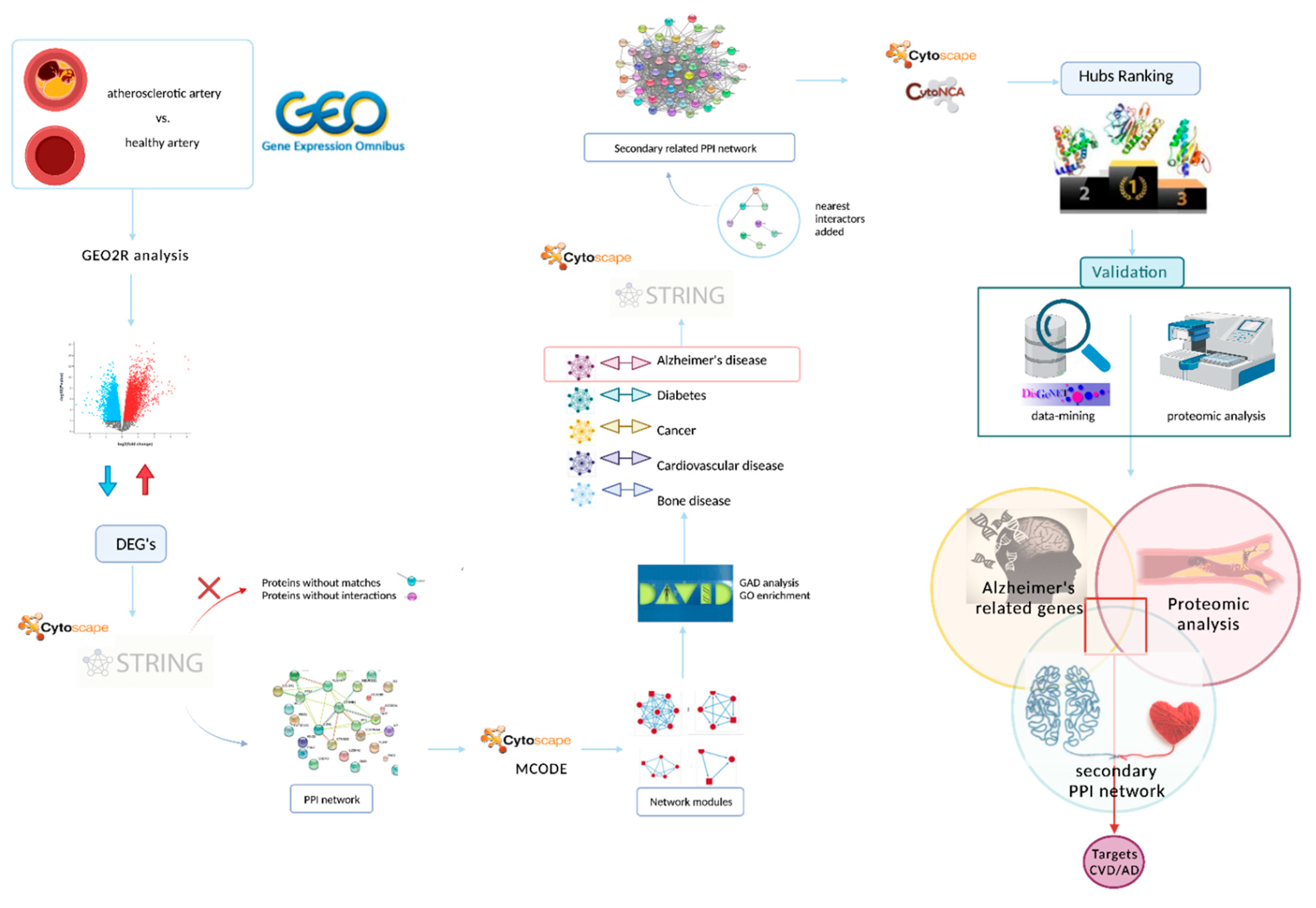
2. Materials and Methods
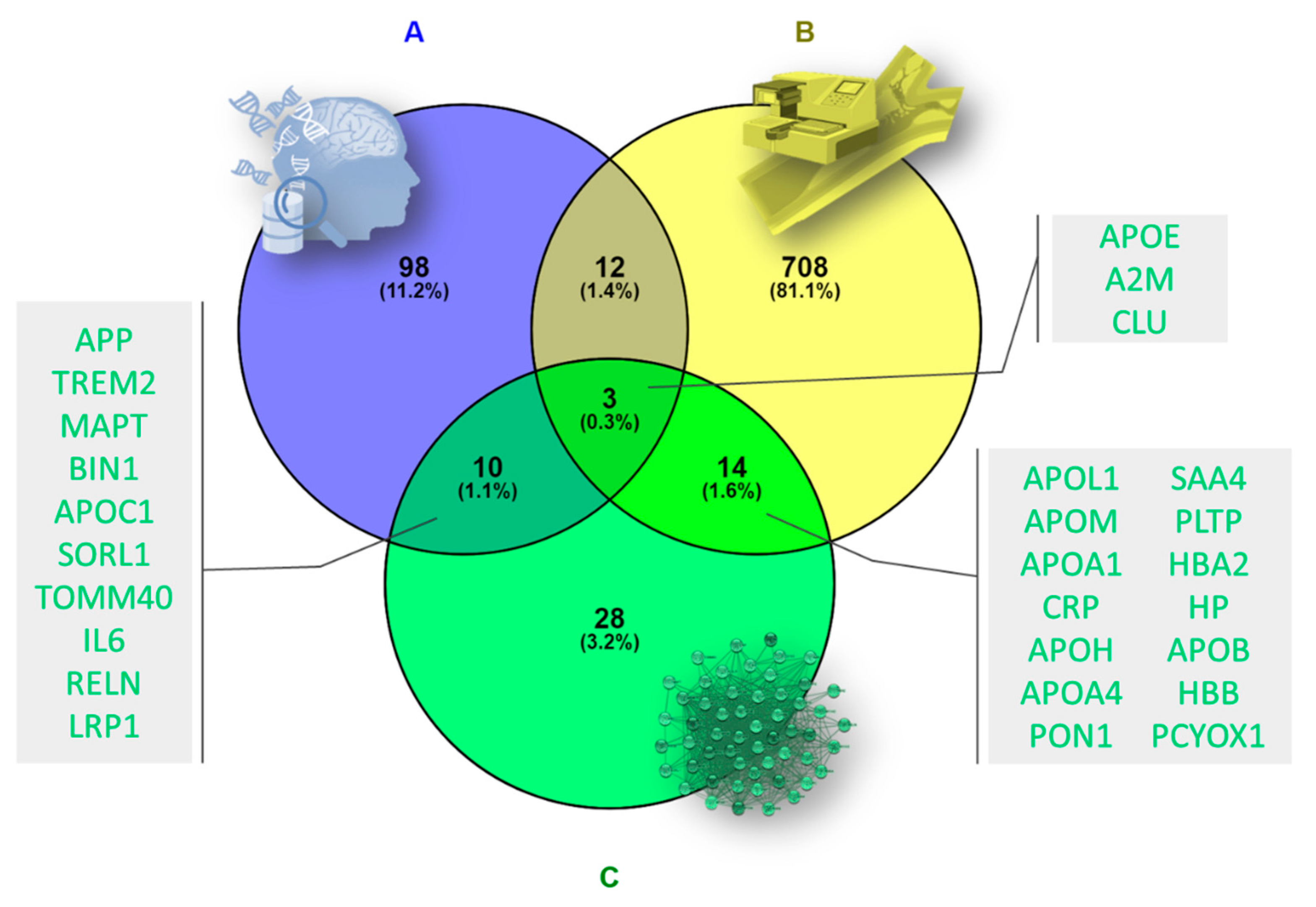
2.1. Data Acquisition
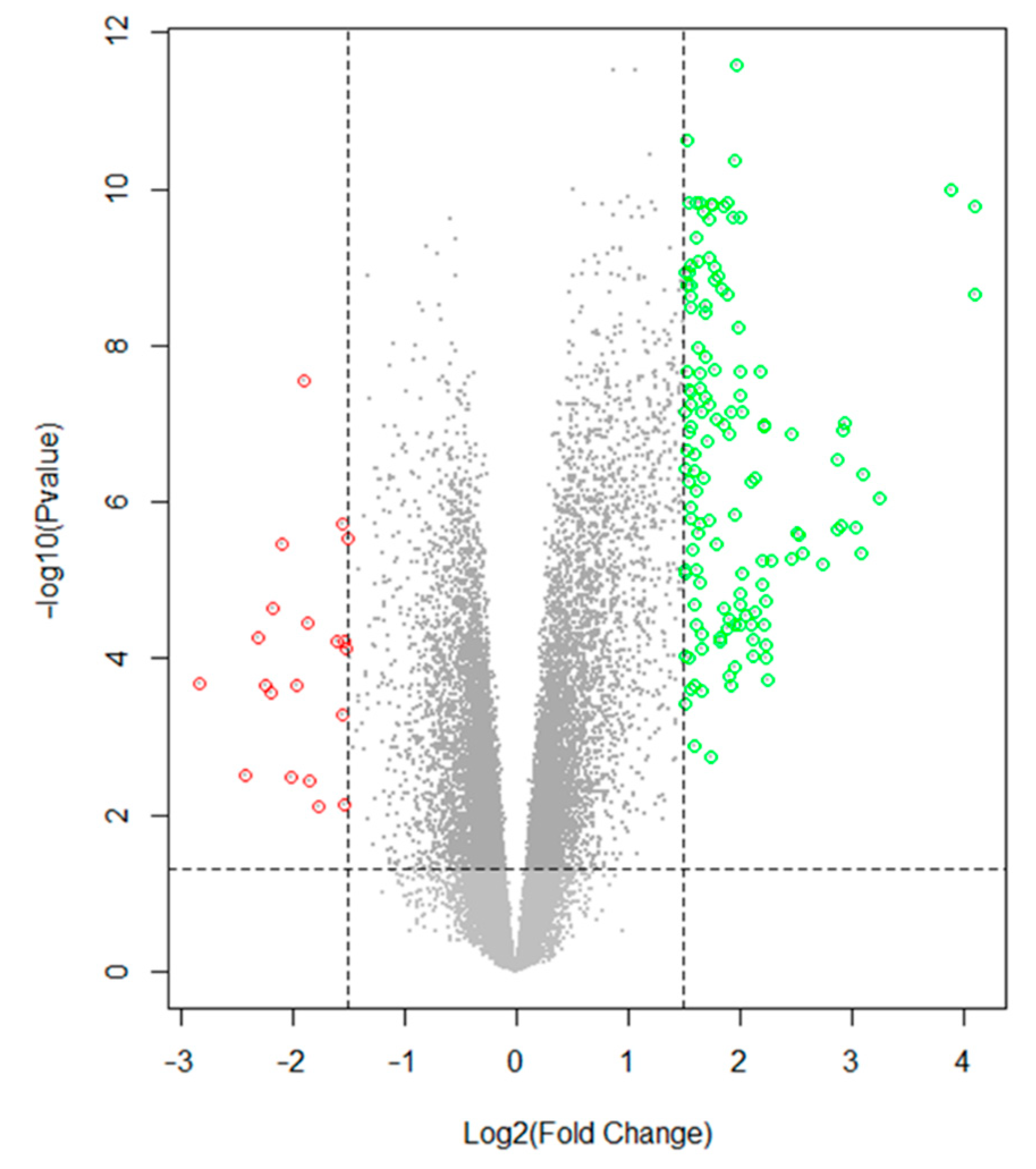
2.2. Data Preprocessing and Identification of Differentially Expressed Genes (DEGs)
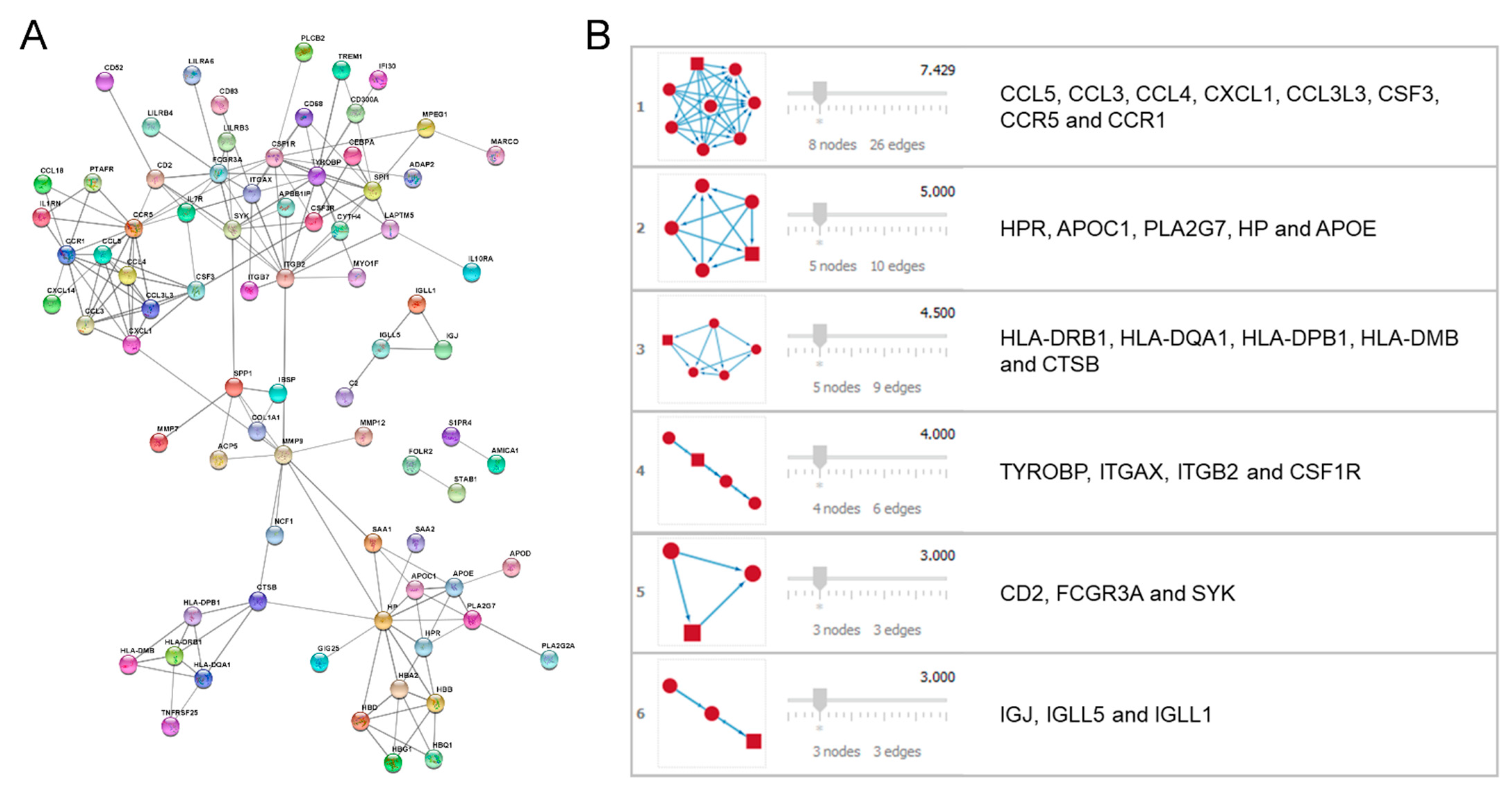
2.3. Protein–Protein Interaction (PPI) Network Performance and Module Analysis
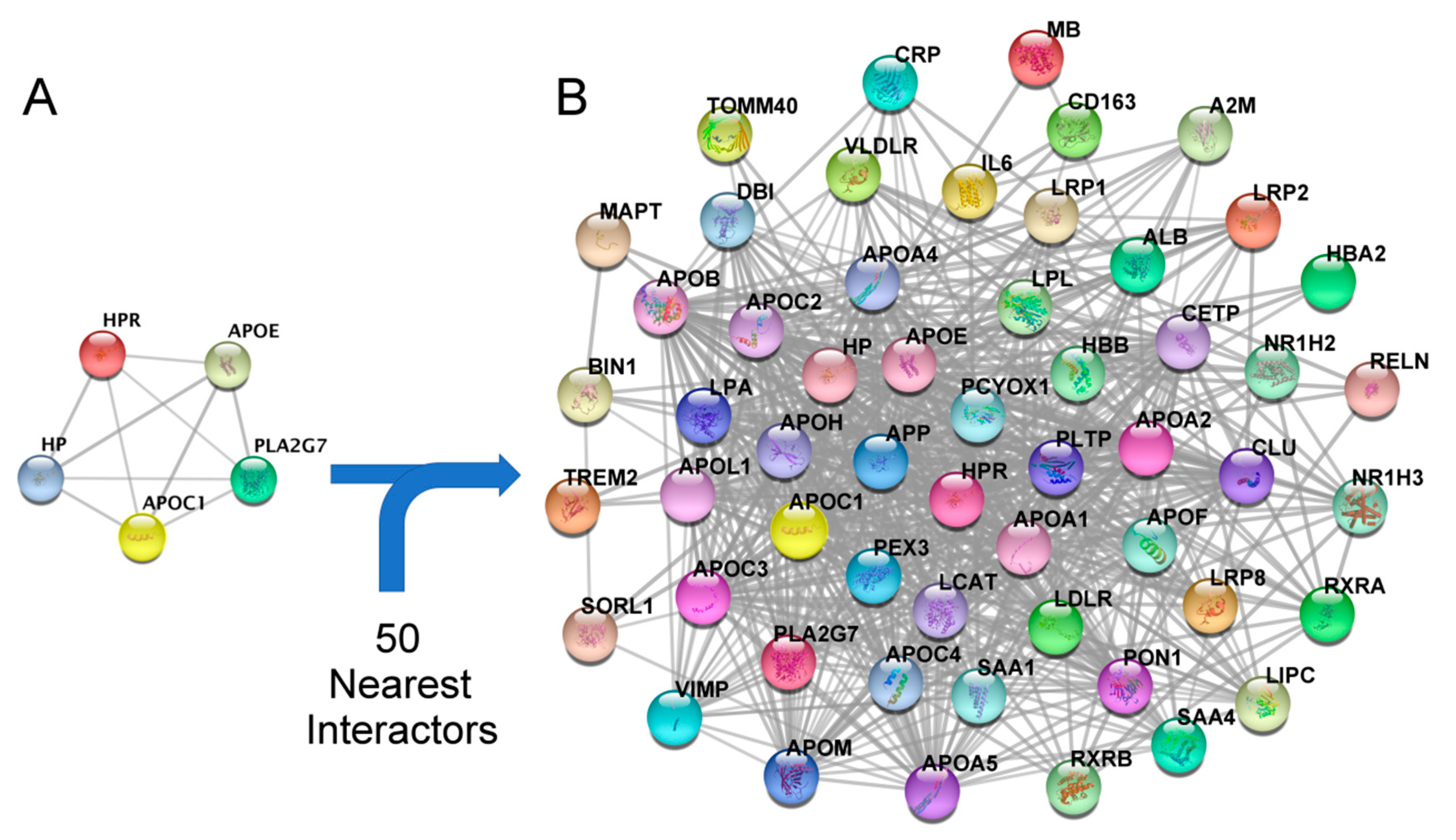
2.4. Construction of a Secondary Related PPI Network and Identification of Hubs
2.5. Validation of Target Proteins by Data-Mining and Proteomic Analysis
3. Results
3.1. Data Acquisition
3.2. Data Preprocessing and Identification of DEGs
3.3. PPI Network Performance and Module Analysis
3.4. Enrichment of the Secondary PPI Network and Identification of Hubs
3.5. Validation of Target Proteins by Data-Mining and Proteomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewis, J.R.; Eggermont, C.J.; Schousboe, J.T.; Lim, W.H.; Wong, G.; Khoo, B.; Sim, M.; Yu, M.; Ueland, T.; Bollerslev, J.; et al. Association Between Abdominal Aortic Calcification, Bone Mineral Density, and Fracture in Older Women. J. Bone Min. Res. 2019, 34, 2052–2060. [Google Scholar] [CrossRef]
- Park, J.; Yoon, Y.E.; Kim, K.M.; Hwang, I.-C.; Lee, W.; Cho, G.-Y. Prognostic Value of Lower Bone Mineral Density in Predicting Adverse Cardiovascular Disease in Asian Women. Heart 2021, 107, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Torres, M.; Reyes-García, R.; García-Martin, A.; Jiménez-Moleón, J.J.; Gonzalez-Ramírez, A.R.; Lara-Villoslada, M.J.; Moreno, P.R. Ischemic Heart Disease Is Associated with Vertebral Fractures in Patients with Type 2 Diabetes Mellitus. J. Diabetes Investig. 2013, 4, 310–315. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Ebner, N.; Dos Santos, M.R.; Ishida, J.; Hasenfuss, G.; von Haehling, S. Cachexia, Muscle Wasting, and Frailty in Cardiovascular Disease. Eur. J. Heart Fail. 2020, 22, 2314–2326. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, A. Roles of Sleep Deprivation in Cardiovascular Dysfunctions. Life Sci. 2019, 219, 231–237. [Google Scholar] [CrossRef]
- Pluta, R. Brain Ischemia as a Bridge to Alzheimer’s Disease. Neural Regen Res. 2021, 17, 791–792. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines; World Health Organization: Geneva, Switzerland, 1992. [Google Scholar]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the Global Burden of Alzheimer’s Disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical Epidemiology of Alzheimer’s Disease: Assessing Sex and Gender Differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Chang, S.M.; Jang, H.-S.; Chang, J.S.; Suh, G.-H.; Jung, H.-Y.; Jeon, H.-J.; Cho, M.J. Illiteracy and the Incidence of Alzheimer’s Disease in the Yonchon County Survey, Korea. Int. Psychogeriatr. 2008, 20, 976–985. [Google Scholar] [CrossRef]
- Cataldo, J.K.; Prochaska, J.J.; Glantz, S.A. Cigarette Smoking Is a Risk Factor for Alzheimer’s Disease: An Analysis Controlling for Tobacco Industry Affiliation. J. Alzheimers Dis. 2010, 19, 465–480. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Wang, Y. Obesity and Central Obesity as Risk Factors for Incident Dementia and Its Subtypes: A Systematic Review and Meta-Analysis. Obes. Rev. 2008, 9, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.-L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Menta, B.W.; Swerdlow, R.H. An Integrative Overview of Non-Amyloid and Non-Tau Pathologies in Alzheimer’s Disease. Neurochem. Res. 2019, 44, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Stamatelopoulos, K.; Bampatsias, D.; Sachse, M.; Zormpas, E.; Vlachogiannis, N.I.; Tual-Chalot, S.; Stellos, K. The Alzheimer’s Disease Amyloid-Beta Hypothesis in Cardiovascular Aging and Disease: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 952–967. [Google Scholar] [CrossRef]
- Wanleenuwat, P.; Iwanowski, P.; Kozubski, W. Alzheimer’s Dementia: Pathogenesis and Impact of Cardiovascular Risk Factors on Cognitive Decline. Postgrad. Med. 2019, 131, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The Pathology and Pathophysiology of Vascular Dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef]
- Iadecola, C. The Pathobiology of Vascular Dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk Factors for Alzheimer Disease. Factores de Riesgo Para La Enfermedad de Alzheimer. Brain Nerve 2019, 57, 87–105. [Google Scholar]
- Kalaria, R.N.; Hedera, P. Differential Degeneration of the Cerebral Microvasculature in Alzheimer’s Disease. NeuroRep. 1995, 6, 477–480. [Google Scholar] [CrossRef]
- Lamar, M.; Boots, E.A.; Arfanakis, K.; Barnes, L.L.; Schneider, J.A. Common Brain Structural Alterations Associated with Cardiovascular Disease Risk Factors and Alzheimer’s Dementia: Future Directions and Implications. Neuropsychol. Rev. 2020, 30, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G. Resolving Atherosclerosis and Alzheimer Disease. Nat. Rev. Cardiol. 2019, 16, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Ruan, J.; Zhang, W. Variations in the Transcriptome of Alzheimer’s Disease Reveal Molecular Networks Involved in Cardiovascular Diseases. Genome Biol. 2008, 9, R148. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Nicolotti, O.; Altomare, C.D. First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules 2021, 26, 5208. [Google Scholar] [CrossRef]
- Henderson, A.S. The Risk Factors for Alzheimer’s Disease: A Review and a Hypothesis. Acta Psychiatr. Scand. 1988, 78, 257–275. [Google Scholar] [CrossRef]
- Hjelm, C.; Broström, A.; Dahl, A.; Johansson, B.; Fredrikson, M.; Strömberg, A. Factors Associated with Increased Risk for Dementia in Individuals Age 80 Years or Older with Congestive Heart Failure. J. Cardiovasc. Nurs. 2014, 29, 82–90. [Google Scholar] [CrossRef]
- Zhang, X.; Le, W. Pathological Role of Hypoxia in Alzheimer’s Disease. Exp. Neurol. 2010, 223, 299–303. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets--Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A Cytoscape Plugin for Centrality Analysis and Evaluation of Protein Interaction Networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A Comprehensive Platform Integrating Information on Human Disease-Associated Genes and Variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.R.; TASC II Working Group. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45 (Suppl. S), S5–S67. [Google Scholar] [CrossRef]
- Andújar-Vera, F.; García-Fontana, C.; Lozano-Alonso, S.; González-Salvatierra, S.; Iglesias-Baena, I.; Muñoz-Torres, M.; García-Fontana, B. Association between Oxidative-Stress-Related Markers and Calcified Femoral Artery in Type 2 Diabetes Patients. J. Pharm. Biomed. Anal. 2020, 190, 113535. [Google Scholar] [CrossRef]
- Steenman, M.; Espitia, O.; Maurel, B.; Guyomarch, B.; Heymann, M.-F.; Pistorius, M.-A.; Ory, B.; Heymann, D.; Houlgatte, R.; Gouëffic, Y.; et al. Identification of Genomic Differences among Peripheral Arterial Beds in Atherosclerotic and Healthy Arteries. Sci. Rep. 2018, 8, 3940. [Google Scholar] [CrossRef]
- Valanti, E.-K.; Dalakoura-Karagkouni, K.; Siasos, G.; Kardassis, D.; Eliopoulos, A.G.; Sanoudou, D. Advances in Biological Therapies for Dyslipidemias and Atherosclerosis. Metabolism 2021, 116, 154461. [Google Scholar] [CrossRef]
- Lee, S.; Parekh, T.; King, S.M.; Reed, B.; Chui, H.C.; Krauss, R.M.; Yassine, H.N. Low-Density Lipoprotein Particle Size Subfractions and Cerebral Amyloidosis. J. Alzheimers Dis. 2019, 68, 983–990. [Google Scholar] [CrossRef]
- Chatterjee, P.; Lim, W.L.F.; Shui, G.; Gupta, V.B.; James, I.; Fagan, A.M.; Xiong, C.; Sohrabi, H.R.; Taddei, K.; Brown, B.M.; et al. Plasma Phospholipid and Sphingolipid Alterations in Presenilin1 Mutation Carriers: A Pilot Study. J. Alzheimers Dis. 2016, 50, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I Deficiency Increases Cortical Amyloid Deposition, Cerebral Amyloid Angiopathy, Cortical and Hippocampal Astrogliosis, and Amyloid-Associated Astrocyte Reactivity in APP/PS1 Mice. Alzheimers Res. 2019, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Tang, Y.; Zhao, L.; Wang, Q.; Qin, W.; Zhang, J.-L.; Jia, J. Plasma Ceramides and Neuropsychiatric Symptoms of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 52, 1029–1035. [Google Scholar] [CrossRef]
- Kim, S.H.; Yang, J.S.; Lee, J.C.; Lee, J.-Y.; Lee, J.-Y.; Kim, E.; Moon, M.H. Lipidomic Alterations in Lipoproteins of Patients with Mild Cognitive Impairment and Alzheimer’s Disease by Asymmetrical Flow Field-Flow Fractionation and Nanoflow Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A. 2018, 1568, 91–100. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of Blood Lipids with Alzheimer’s Disease: A Comprehensive Lipidomics Analysis. Alzheimers Dement. 2017, 13, 140–151. [Google Scholar] [CrossRef]
- Mejías-Trueba, M.; Pérez-Moreno, M.A.; Fernández-Arche, M.Á. Systematic Review of the Efficacy of Statins for the Treatment of Alzheimer’s Disease. Clin. Med. Lond. 2018, 18, 54–61. [Google Scholar] [CrossRef]
- Chaaba, R.; Attia, N.; Hammami, S.; Smaoui, M.; Ben Hamda, K.; Mahjoub, S.; Hammami, M. Association between Apolipoprotein E Polymorphism, Lipids, and Coronary Artery Disease in Tunisian Type 2 Diabetes. J. Clin. Lipidol. 2008, 2, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Fu, X.; Chu, Q.; Li, J.; Shu, G. Relationship Between the ApoE Gene Polymorphism and Type 2 Diabetes Mellitus Complications. Genet. Test. Mol. Biomark. 2021, 25, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-Q.; Ren, H.; Banh, H.L.; Liu, M.-Z.; Xu, P.; Fang, P.-F.; Xiang, D.-X. The Associations between Apolipoprotein E Gene Epsilon2/Epsilon3/Epsilon4 Polymorphisms and the Risk of Coronary Artery Disease in Patients with Type 2 Diabetes Mellitus. Front Physiol. 2017, 8, 1031. [Google Scholar] [CrossRef] [PubMed]
- Borovecki, F.; Klepac, N.; Muck-Seler, D.; Hajnsek, S.; Mubrin, Z.; Pivac, N. Unraveling the Biological Mechanisms in Alzheimer’s Disease--Lessons from Genomics. Prog Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 340–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.-C.; Chiu, Y.-J.; Lin, C.-H.; Hsu, W.-C.; Wu, J.-L.; Huang, C.-H.; Lin, C.-W.; Yao, C.-F.; Huang, H.-J.; Lo, Y.-S.; et al. Indole Compound NC009-1 Augments APOE and TRKA in Alzheimer’s Disease Cell and Mouse Models for Neuroprotection and Cognitive Improvement. J. Alzheimers Dis. 2019, 67, 737–756. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Marks, J.D.; Fan, Z.; Klickstein, J.A.; Roe, A.D.; Krogh, K.A.; Wegmann, S.; Maesako, M.; Luo, C.C.; Mylvaganam, R.; et al. Isoform- and Cell Type-Specific Structure of Apolipoprotein E Lipoparticles as Revealed by a Novel Forster Resonance Energy Transfer Assay. J. Biol. Chem. 2017, 292, 14720–14729. [Google Scholar] [CrossRef] [PubMed]
- McFall, G.P.; Bäckman, L.; Dixon, R.A. Nuances in Alzheimer’s Genetic Risk Reveal Differential Predictions of Non-Demented Memory Aging Trajectories: Selective Patterns by APOE Genotype and Sex. Curr. Alzheimer Res. 2019, 16, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Utermann, G. Alzheimer’s Disease. The Apolipoprotein E Connection. Curr. Biol. 1994, 4, 362–365. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer Disease: Pathobiology and Targeting Strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Marais, A.D. Apolipoprotein E in Lipoprotein Metabolism, Health and Cardiovascular Disease. Patholology 2019, 51, 165–176. [Google Scholar] [CrossRef]
- Borth, W. Alpha 2-Macroglobulin, a Multifunctional Binding Protein with Targeting Characteristics. FASEB J. 1992, 6, 3345–3353. [Google Scholar] [CrossRef]
- Varma, V.R.; Varma, S.; An, Y.; Hohman, T.J.; Seddighi, S.; Casanova, R.; Beri, A.; Dammer, E.B.; Seyfried, N.T.; Pletnikova, O.; et al. Alpha-2 Macroglobulin in Alzheimer’s Disease: A Marker of Neuronal Injury through the RCAN1 Pathway. Mol. Psychiatry 2017, 22, 13–23. [Google Scholar] [CrossRef]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular Form and Concentration of Serum A2-Macroglobulin in Diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef]
- Nezu, T.; Hosomi, N.; Aoki, S.; Deguchi, K.; Masugata, H.; Ichihara, N.; Ohyama, H.; Ohtsuki, T.; Kohno, M.; Matsumoto, M. Alpha2-Macroglobulin as a Promising Biomarker for Cerebral Small Vessel Disease in Acute Ischemic Stroke Patients. J. Neurol. 2013, 260, 2642–2649. [Google Scholar] [CrossRef]
- Jones, S.E.; Jomary, C. Clusterin. Int. J. Biochem. Cell Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]
- Wong, P.; Taillefer, D.; Lakins, J.; Pineault, J.; Chader, G.; Tenniswood, M. Molecular Characterization of Human TRPM-2/Clusterin, a Gene Associated with Sperm Maturation, Apoptosis and Neurodegeneration. Eur. J. Biochem. 1994, 221, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Al Nakouzi, N.; Wang, C.K.; Beraldi, E.; Jager, W.; Ettinger, S.; Fazli, L.; Nappi, L.; Bishop, J.; Zhang, F.; Chauchereau, A.; et al. Clusterin Knockdown Sensitizes Prostate Cancer Cells to Taxane by Modulating Mitosis. EMBO Mol. Med. 2016, 8, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Tsai, P.; Sun, H.-Y.; Hsu, M.-C.; Lee, J.-C.; Wu, I.-C.; Tsao, C.-W.; Chang, T.-T.; Young, K.-C. Apolipoprotein, J., A Glucose-Upregulated Molecular Chaperone, Stabilizes Core and NS5A to Promote Infectious Hepatitis C Virus Virion Production. J. Hepatol. 2014, 61, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Gonos, E.S. Regulation of Clusterin/Apolipoprotein J, a Functional Homologue to the Small Heat Shock Proteins, by Oxidative Stress in Ageing and Age-Related Diseases. Free Radic. Res. 2006, 40, 1324–1334. [Google Scholar] [CrossRef]
- Wong, P.; Pineault, J.; Lakins, J.; Taillefer, D.; Léger, J.; Wang, C.; Tenniswood, M. Genomic Organization and Expression of the Rat TRPM-2 (Clusterin) Gene, a Gene Implicated in Apoptosis. J. Biol. Chem. 1993, 268, 5021–5031. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef]
- May, P.C.; Finch, C.E. Sulfated Glycoprotein 2: New Relationships of This Multifunctional Protein to Neurodegeneration. Trends Neurosci. 1992, 15, 391–396. [Google Scholar] [CrossRef]
- Roheim, P.S.; Carey, M.; Forte, T.; Vega, G.L. Apolipoproteins in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 1979, 76, 4646–4649. [Google Scholar] [CrossRef]
- May, P.C.; Lampert-Etchells, M.; Johnson, S.A.; Poirier, J.; Masters, J.N.; Finch, C.E. Dynamics of Gene Expression for a Hippocampal Glycoprotein Elevated in Alzheimer’s Disease and in Response to Experimental Lesions in Rat. Neuron 1990, 5, 831–839. [Google Scholar] [CrossRef]
- Calero, M.; Rostagno, A.; Matsubara, E.; Zlokovic, B.; Frangione, B.; Ghiso, J. Apolipoprotein J (Clusterin) and Alzheimer’s Disease. Microsc. Res. Tech. 2000, 50, 305–315. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Martel, C.L.; Matsubara, E.; McComb, J.G.; Zheng, G.; McCluskey, R.T.; Frangione, B.; Ghiso, J. Glycoprotein 330/Megalin: Probable Role in Receptor-Mediated Transport of Apolipoprotein J Alone and in a Complex with Alzheimer Disease Amyloid Beta at the Blood-Brain and Blood-Cerebrospinal Fluid Barriers. Proc. Natl. Acad. Sci. USA 1996, 93, 4229–4234. [Google Scholar] [CrossRef]
- Ghiso, J.; Matsubara, E.; Koudinov, A.; Choi-Miura, N.H.; Tomita, M.; Wisniewski, T.; Frangione, B. The Cerebrospinal-Fluid Soluble Form of Alzheimer’s Amyloid β Is Complexed to SP-40,40 (Apolipoprotein J), an Inhibitor of the Complement Membrane-Attack Complex. Biochem. J. 1993, 293, 27–30. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Cirrito, J.R.; Parsadanian, M.; May, P.C.; O’Dell, M.A.; Taylor, J.W.; Harmony, J.A.K.; Aronow, B.J.; Bales, K.R.; Paul, S.M.; et al. ApoE and Clusterin Cooperatively Suppress Aβ Levels and Deposition: Evidence That ApoE Regulates Extracellular Aβ Metabolism In Vivo. Neuron 2004, 41, 193–202. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holtzman, D.M.; Deane, R.; Zlokovic, B.V. Transport Pathways for Clearance of Human Alzheimer’s Amyloid β-Peptide and Apolipoproteins E and J in the Mouse Central Nervous System. J. Cereb. Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Shi, Y.; Yuan, Y.; Xie, C.; Zhang, Z. Immunity Factor Contributes to Altered Brain Functional Networks in Individuals at Risk for Alzheimer’s Disease: Neuroimaging-Genetic Evidence. Brain Behav. Immun. 2016, 56, 84–95. [Google Scholar] [CrossRef]
- Schrijvers, E.M.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Plasma Clusterin and the Risk of Alzheimer Disease. JAMA 2011, 305, 1322–1326. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Li, C.; Niu, H.; Luo, S.; Guo, X. Association between Clusterin Concentration and Dementia: A Systematic Review and Meta-Analysis. Metab. Brain Dis. 2019, 34, 129–140. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Lee, W.-J.; Liao, Y.-C.; Wang, S.-J.; Fuh, J.-L. The Clinical Significance of Plasma Clusterin and Aβ in the Longitudinal Follow-up of Patients with Alzheimer’s Disease. Alzheimers Res. 2017, 9, 91. [Google Scholar] [CrossRef]
- Bradley, D.; Blaszczak, A.; Yin, Z.; Liu, J.; Joseph, J.J.; Wright, V.; Anandani, K.; Needleman, B.; Noria, S.; Renton, D.; et al. Clusterin Impairs Hepatic Insulin Sensitivity and Adipocyte Clusterin Associates With Cardiometabolic Risk. Diabetes Care 2019, 42, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Burk, D.H.; Ali, M.R.; Mostaedi, R.; Smith, W.H.; Park, J.; Scherer, P.E.; Seay, S.A.; McCoin, C.S.; Bonaldo, P.; et al. Contributions of Adipose Tissue Architectural and Tensile Properties toward Defining Healthy and Unhealthy Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E233–E246. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Chun, T.-H.; Kang, L. Adipose Extracellular Matrix Remodelling in Obesity and Insulin Resistance. Biochem. Pharm. 2016, 119, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Won, J.C.; Park, C.-Y.; Oh, S.W.; Lee, E.S.; Youn, B.-S.; Kim, M.-S. Plasma Clusterin (ApoJ) Levels Are Associated with Adiposity and Systemic Inflammation. PLoS ONE 2014, 9, e103351. [Google Scholar] [CrossRef]
- Kim, H.-J.; Yoo, E.-K.; Kim, J.-Y.; Choi, Y.-K.; Lee, H.-J.; Kim, J.-K.; Jeoung, N.H.; Lee, K.-U.; Park, I.-S.; Min, B.-H.; et al. Protective Role of Clusterin/Apolipoprotein J against Neointimal Hyperplasia via Antiproliferative Effect on Vascular Smooth Muscle Cells and Cytoprotective Effect on Endothelial Cells. Arter. Thromb Vasc. Biol. 2009, 29, 1558–1564. [Google Scholar] [CrossRef]
- Baralla, A.; Sotgiu, E.; Deiana, M.; Pasella, S.; Pinna, S.; Mannu, A.; Canu, E.; Sotgiu, G.; Ganau, A.; Zinellu, A.; et al. Plasma Clusterin and Lipid Profile: A Link with Aging and Cardiovascular Diseases in a Population with a Consistent Number of Centenarians. PLoS ONE 2015, 10, e0128029. [Google Scholar] [CrossRef]
- Park, S.; Mathis, K.W.; Lee, I.K. The Physiological Roles of Apolipoprotein J/Clusterin in Metabolic and Cardiovascular Diseases. Rev. Endocr. Metab. Disord. 2014, 15, 45–53. [Google Scholar] [CrossRef]
- Wittwer, J.; Bradley, D. Clusterin and Its Role in Insulin Resistance and the Cardiometabolic Syndrome. Front Immunol. 2021, 12, 612496. [Google Scholar] [CrossRef]
- Güleç, G.U.; Turgut, Y.B.; Turgut, M. Acute phase proteins. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 978-0-12-801238-3. [Google Scholar]
- Song, I.-U.; Kim, Y.-D.; Chung, S.-W.; Cho, H.-J. Association between Serum Haptoglobin and the Pathogenesis of Alzheimer’s Disease. Intern. Med. 2015, 54, 453–457. [Google Scholar] [CrossRef]
- Zhu, C.-J.; Jiang, G.-X.; Chen, J.-M.; Zhou, Z.-M.; Cheng, Q. Serum Haptoglobin in Chinese Patients with Alzheimer’s Disease and Mild Cognitive Impairment: A Case-Control Study. Brain Res. Bull. 2018, 137, 301–305. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | % | p-Value | Genes |
|---|---|---|---|---|
| GAD_DISEASE | cardiovascular disease | 80 | 3.56 × 10−7 | APOC1, HP, APOE, PLA2G7 |
| GAD_DISEASE | atherosclerosis, coronary | 80 | 6.64 × 10−6 | APOC1, HP, APOE, PLA2G7 |
| GOTERM_CC_DIRECT | GO:0005576~extracellular region | 100 | 6.07 × 10−5 | APOC1, HP, HPR, APOE, PLA2G7 |
| GAD_DISEASE | cholesterol | 80 | 1.38 × 10−4 | APOC1, HPR, APOE, PLA2G7 |
| GAD_DISEASE | coronary disease; coronary heart disease | 60 | 1.61 × 10−4 | APOC1, APOE, PLA2G7 |
| GOTERM_CC_DIRECT | GO:0072562~blood microparticle | 60 | 4.10 × 10−4 | HP, HPR, APOE |
| GAD_DISEASE | familial dysbetalipoproteinemia | 40 | 6.17 × 10−4 | APOC1, APOE |
| GOTERM_BP_DIRECT | GO:0034447~very-low-density lipoprotein particle clearance | 40 | 7.14 × 10−4 | APOC1, APOE |
| GOTERM_BP_DIRECT | GO:0006898~receptor-mediated endocytosis | 60 | 7.22 × 10−4 | HP, HPR, APOE |
| GAD_DISEASE | type 2 diabetes; edema; rosiglitazone | 100 | 8.23 × 10−4 | APOC1, HP, HPR, APOE, PLA2G7 |
| GAD_DISEASE | coronary disease; diabetes complications; hypercholesterolemia; hypertension; myocardial infarction | 40 | 9.25 × 10−4 | APOC1, APOE |
| GOTERM_MF_DIRECT | GO:0030492~hemoglobin binding | 40 | 9.48 × 10−4 | HP, HPR |
| GAD_DISEASE | cholesterol; coronary heart disease; lipoproteins | 40 | 1.23 × 10−3 | APOC1, APOE |
| GAD_DISEASE | cardiovascular diseases | 60 | 1.24 × 10−3 | APOC1, APOE, PLA2G7 |
| GOTERM_MF_DIRECT | GO:0060228~phosphatidylcholine-sterol O-acyltransferase activator activity | 40 | 1.42 × 10−3 | APOC1, APOE |
| GOTERM_BP_DIRECT | GO:0034382~chylomicron remnant clearance | 40 | 1.43 × 10−3 | APOC1, APOE |
| GAD_DISEASE | Alzheimer’s disease | 80 | 1.48 × 10−3 | APOC1, HP, APOE, PLA2G7 |
| GAD_DISEASE | memory disturbance | 40 | 1.85 × 10−3 | APOC1, APOE |
| Symbol | Description | Degree | Betweenness |
|---|---|---|---|
| APOE | Apolipoprotein E | 50.0 | 416.64447 |
| APOA1 | Apolipoprotein A1 | 42.0 | 118.15003 |
| APOC2 | Apolipoprotein C2 | 40.0 | 71.67373 |
| APP | Amyloid Beta Precursor Protein | 39.0 | 196.27913 |
| APOA2 | Apolipoprotein A2 | 39.0 | 57.75081 |
| APOC1 | Apolipoprotein C1 | 39.0 | 97.92878 |
| APOB | Apolipoprotein B | 38.0 | 47.32024 |
| CLU | Clusterin | 37.0 | 84.87135 |
| APOC3 | Apolipoprotein C3 | 37.0 | 36.22977 |
| PLTP | Phospholipid Transfer Protein | 36.0 | 41.08785 |
| CETP | Cholesteryl Ester Transfer Protein | 36.0 | 43.38615 |
| HP | Haptoglobin | 36.0 | 234.20578 |
| APOA4 | Apolipoprotein A4 | 35.0 | 25.99814 |
| APOC4 | Apolipoprotein C4 | 33.0 | 32.60615 |
| APOA5 | Apolipoprotein A5 | 33.0 | 416.64447 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andújar-Vera, F.; García-Fontana, C.; Sanabria-de la Torre, R.; González-Salvatierra, S.; Martínez-Heredia, L.; Iglesias-Baena, I.; Muñoz-Torres, M.; García-Fontana, B. Identification of Potential Targets Linked to the Cardiovascular/Alzheimer’s Axis through Bioinformatics Approaches. Biomedicines 2022, 10, 389. https://doi.org/10.3390/biomedicines10020389
Andújar-Vera F, García-Fontana C, Sanabria-de la Torre R, González-Salvatierra S, Martínez-Heredia L, Iglesias-Baena I, Muñoz-Torres M, García-Fontana B. Identification of Potential Targets Linked to the Cardiovascular/Alzheimer’s Axis through Bioinformatics Approaches. Biomedicines. 2022; 10(2):389. https://doi.org/10.3390/biomedicines10020389
Chicago/Turabian StyleAndújar-Vera, Francisco, Cristina García-Fontana, Raquel Sanabria-de la Torre, Sheila González-Salvatierra, Luis Martínez-Heredia, Iván Iglesias-Baena, Manuel Muñoz-Torres, and Beatriz García-Fontana. 2022. "Identification of Potential Targets Linked to the Cardiovascular/Alzheimer’s Axis through Bioinformatics Approaches" Biomedicines 10, no. 2: 389. https://doi.org/10.3390/biomedicines10020389
APA StyleAndújar-Vera, F., García-Fontana, C., Sanabria-de la Torre, R., González-Salvatierra, S., Martínez-Heredia, L., Iglesias-Baena, I., Muñoz-Torres, M., & García-Fontana, B. (2022). Identification of Potential Targets Linked to the Cardiovascular/Alzheimer’s Axis through Bioinformatics Approaches. Biomedicines, 10(2), 389. https://doi.org/10.3390/biomedicines10020389