
Using the Power of Junctional Adhesion Molecules Combined with the Target of CAR-T to Inhibit Cancer Proliferation, Metastasis and Eradicate Tumors



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cloning, Protein Expression, and Purification
2.2. Size Exclusion Chromatography (SEC)
2.3. SDS-PAGE Assay
2.4. Tissue Culture and In Vitro Experiments with CAL27 and A549 Cells
2.5. Proliferation Assay
2.6. Wound Healing Assay
2.7. Real-Time Cell Invasion
2.8. CAR-T Cell Killing Assay
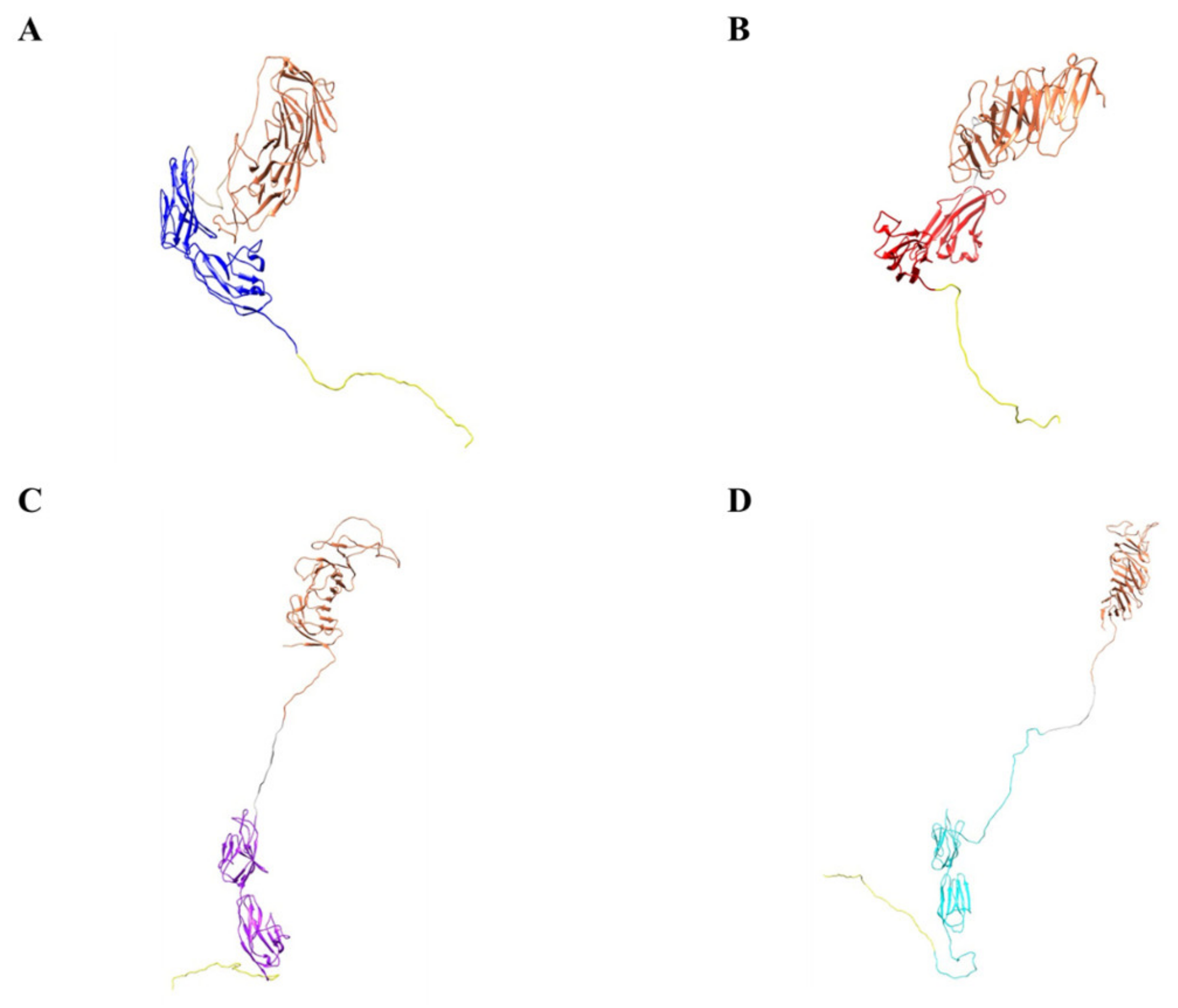
2.9. Computer Models of the Biologic
2.10. Statistical Analysis
3. Results and Discussion
3.1. Expression and Purification of CM19XA in E. coli
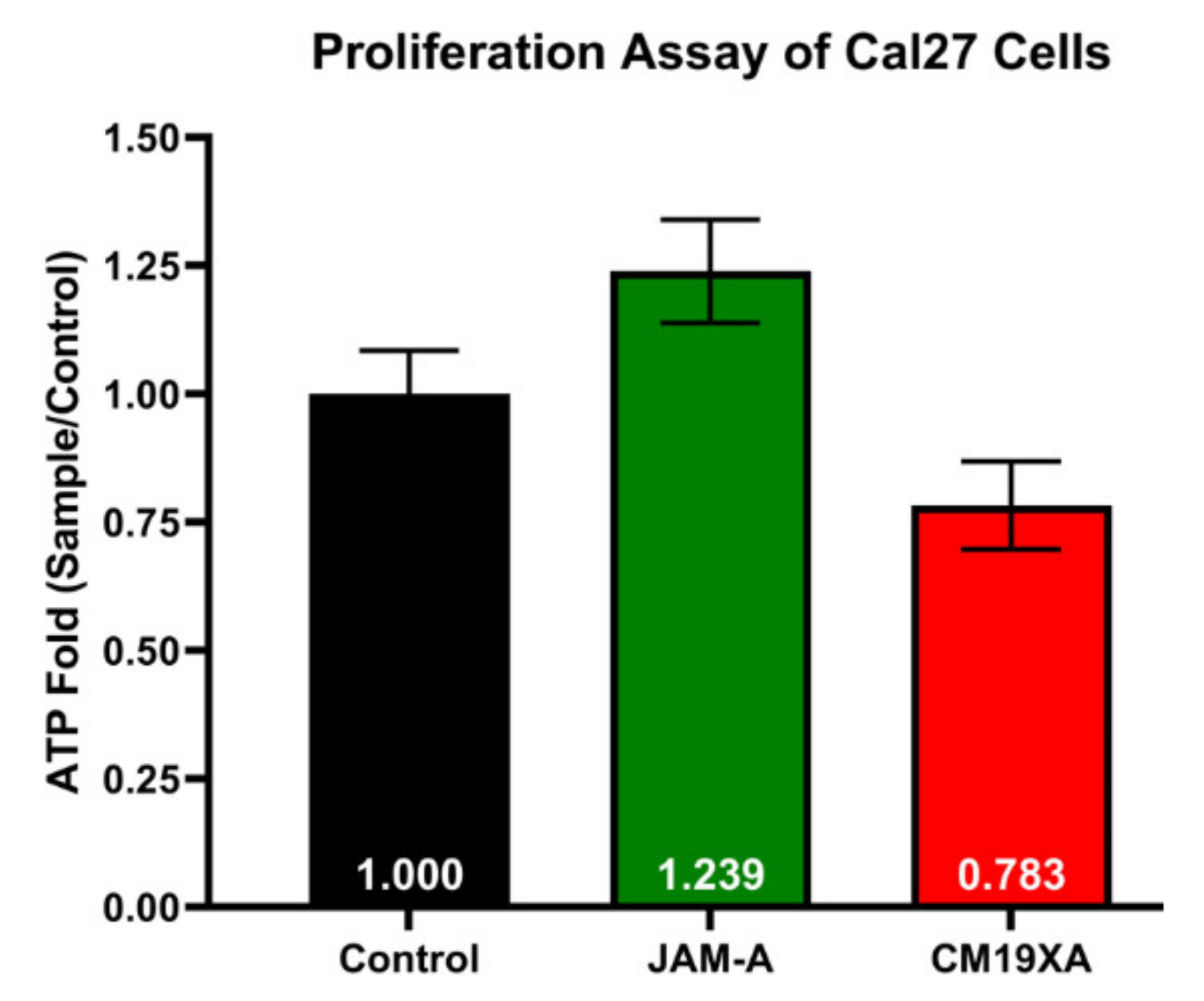
3.2. Proliferation Assay
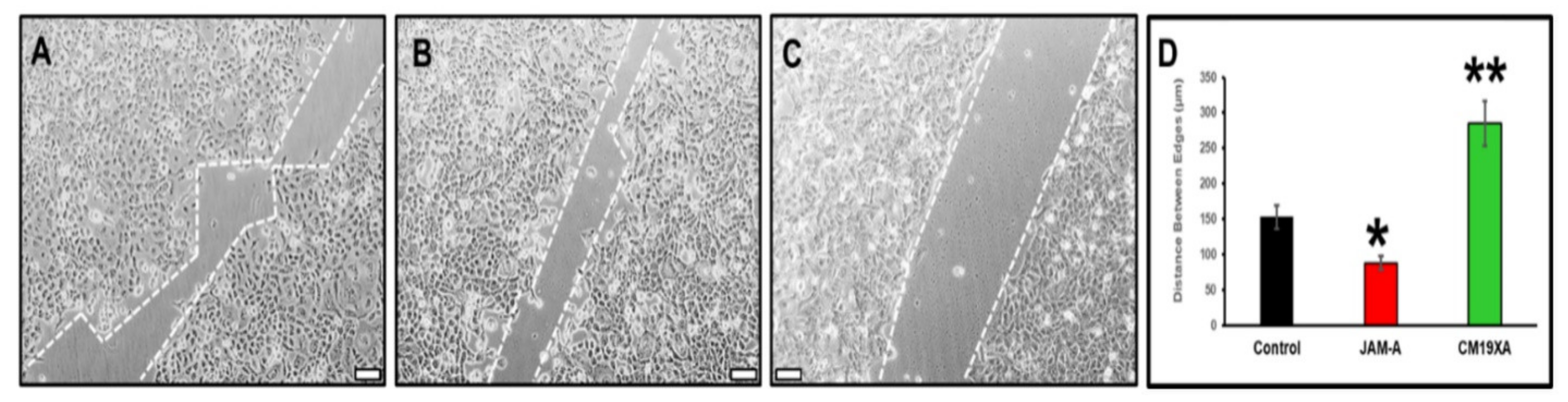
3.3. Wound Healing Assay
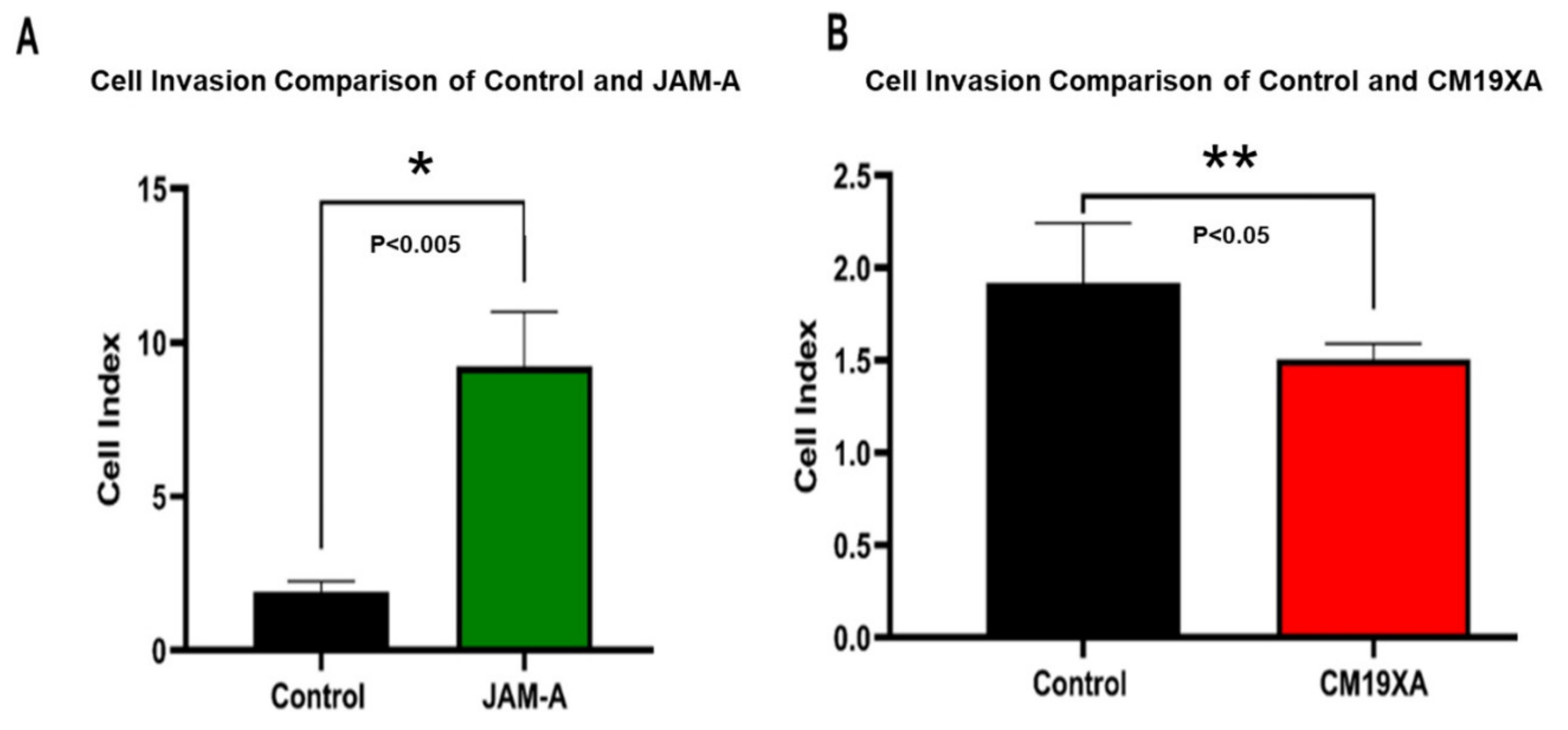
3.4. Cell Invasion Assay
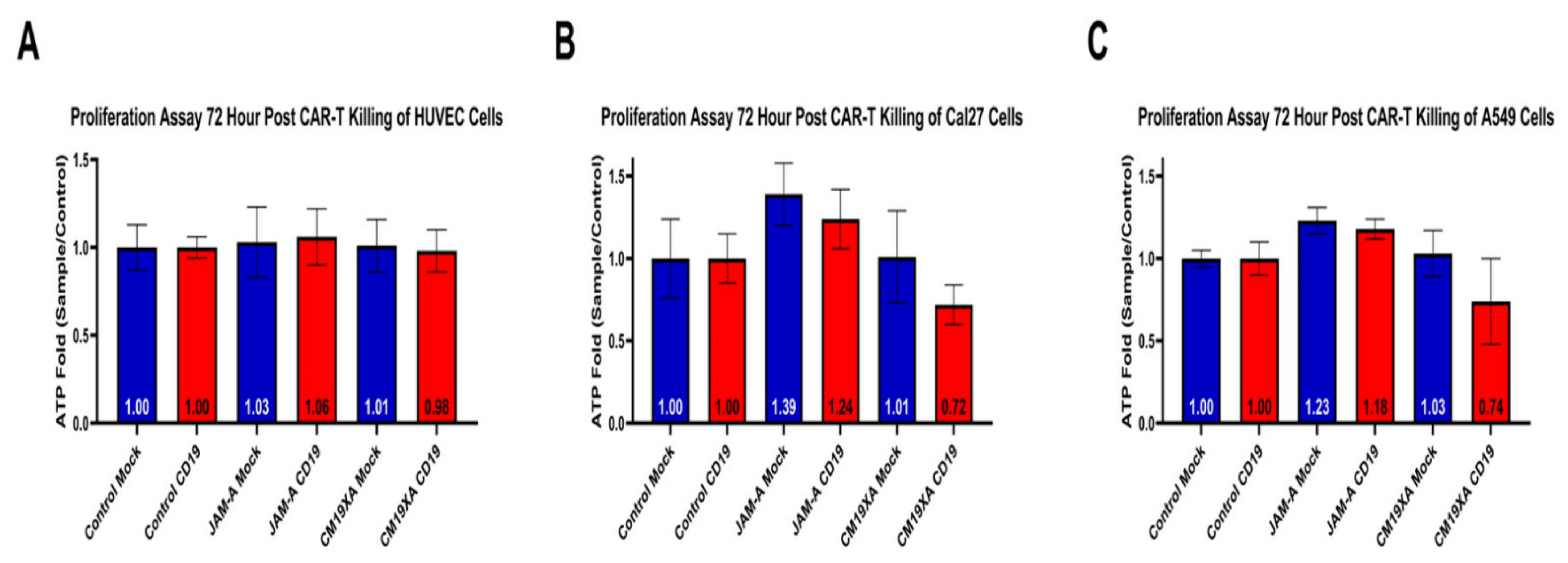
3.5. CM19XA Only Targets Cancer Cells
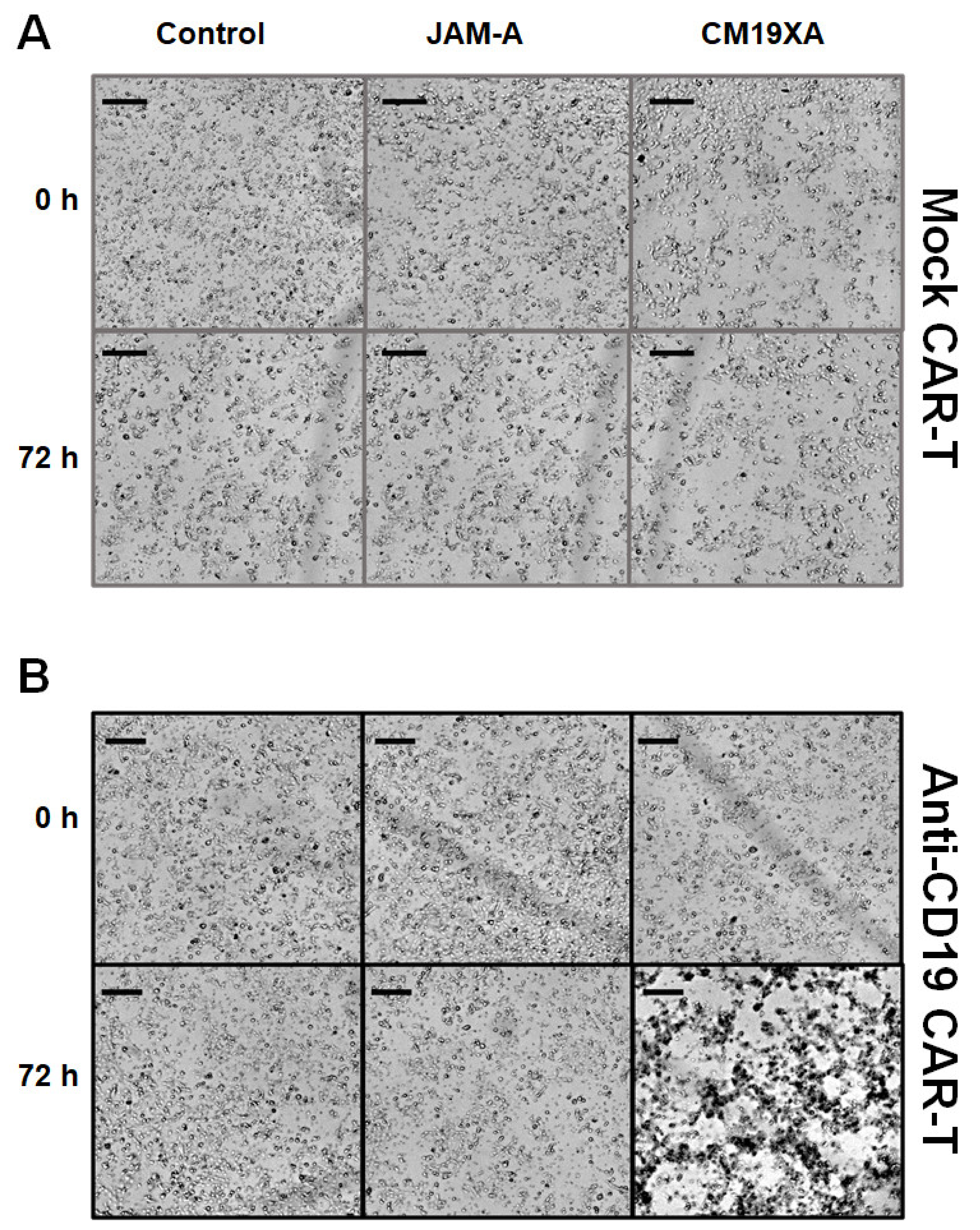
3.6. Cytotoxicity Assay
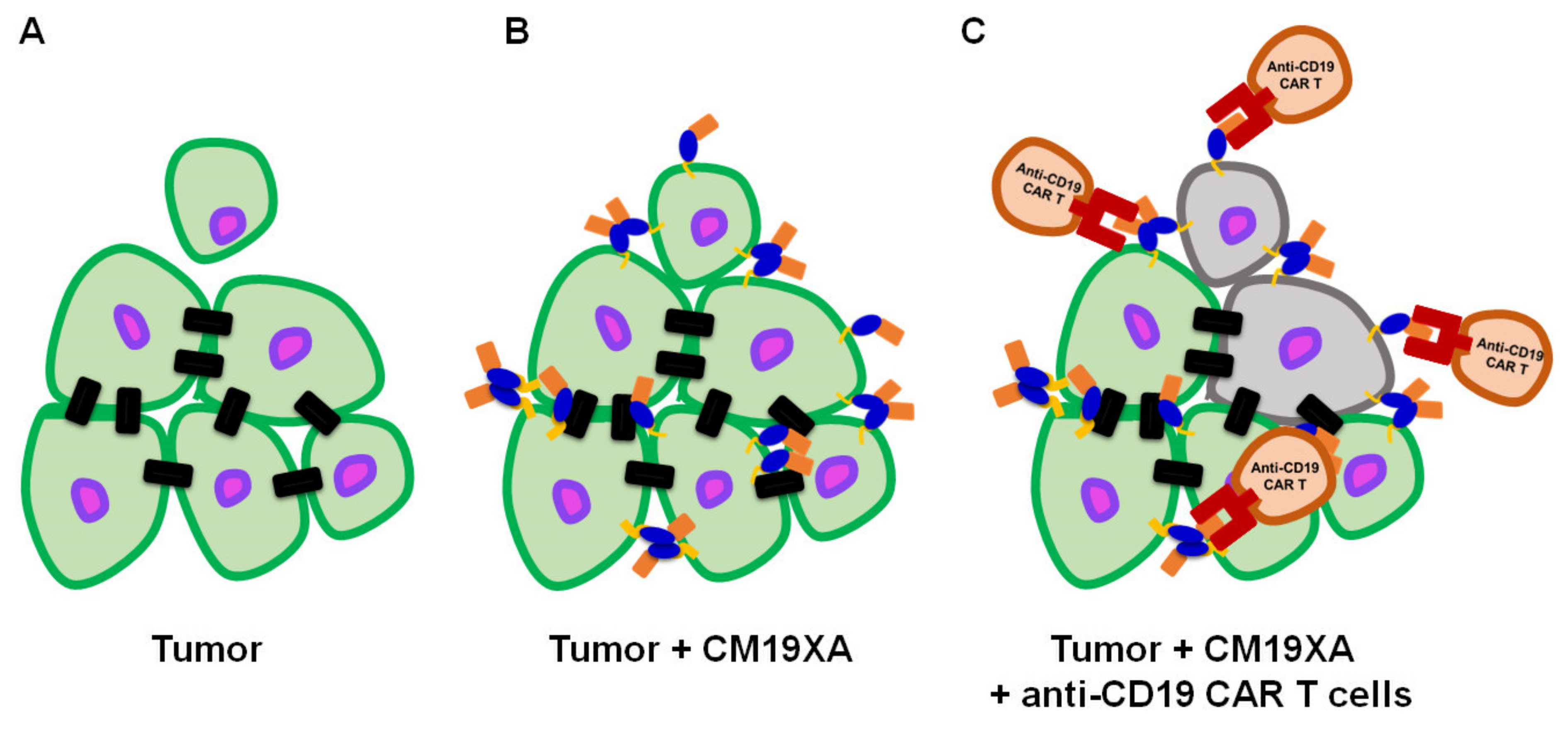
3.7. Proposed Mode of Action of CM19XA
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Shimono, Y.; Rikitake, Y.; Mandai, K.; Mori, M.; Takai, Y. Immunoglobulin superfamily receptors and adherens junctions. Subcell. Biochem. 2012, 60, 137–170. [Google Scholar] [CrossRef] [PubMed]
- Verschueren, E.; Husain, B.; Yuen, K.; Sun, Y.; Paduchuri, S.; Senbabaoglu, Y.; Lehoux, I.; Arena, T.A.; Wilson, B.; Lianoglou, S.; et al. The immunoglobulin superfamily receptome defines cancer-relevant networks associated with clinical outcome. Cell 2020, 182, 329.e319–344.e319. [Google Scholar] [CrossRef]
- Wai Wong, C.; Dye, D.E.; Coombe, D.R. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 2012, 340296. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, T.; Kummer, D.; Ebnet, K. Junctional adhesion molecule-A: Functional diversity through molecular promiscuity. Cell Mol. Life Sci. 2018, 75, 1393–1409. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K. Junctional Adhesion Molecules (JAMs): Cell adhesion receptors with pleiotropic functions in cell physiology and development. Physiol. Rev. 2017, 97, 1529–1554. [Google Scholar] [CrossRef]
- Mendoza, C.; Nagidi, S.H.; Mizrachi, D. Molecular Characterization of the extracellular domain of human junctional adhesion proteins. Int. J. Mol. Sci. 2021, 22, 3482. [Google Scholar] [CrossRef]
- Mendoza, C.; Nagidi, S.H.; Collett, K.; McKell, J.; Mizrachi, D. Calcium regulates the interplay between the tight junction and epithelial adherens junction at the plasma membrane. FEBS Lett. 2021, 596, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Context-dependent roles of claudins in tumorigenesis. Front. Oncol. 2021, 11, 676781. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, J.; Han, Y.; Zhang, Y.; Su, L.; Hu, D.; Fu, X. JAM-A knockdown accelerates the proliferation and migration of human keratinocytes, and improves wound healing in rats via FAK/Erk signaling. Cell Death Dis. 2018, 9, 848. [Google Scholar] [CrossRef]
- Tian, Y.; Tian, Y.; Zhang, W.; Wei, F.; Yang, J.; Luo, X.; Zhou, T.; Hou, B.; Qian, S.; Deng, X.; et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis 2015, 36, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turaga, S.M.; Silver, D.J.; Bayik, D.; Paouri, E.; Peng, S.; Lauko, A.; Alban, T.J.; Borjini, N.; Stanko, S.; Naik, U.P.; et al. JAM-A functions as a female microglial tumor suppressor in glioblastoma. Neuro-Oncol. 2020, 22, 1591–1601. [Google Scholar] [CrossRef]
- Lee, J.K.; Bangayan, N.J.; Chai, T.; Smith, B.A.; Pariva, T.E.; Yun, S.; Vashisht, A.; Zhang, Q.; Park, J.W.; Corey, E.; et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E4473–E4482. [Google Scholar] [CrossRef] [Green Version]
- Tedder, T.F.; Isaacs, C.M. Isolation of cDNAs encodi.ing the CD19 antigen of human and mouse B lymphocytes. A new member of the immunoglobulin superfamily. J. Immunol. 1989, 143, 712–717. [Google Scholar]
- Nadler, L.M.; Anderson, K.C.; Marti, G.; Bates, M.; Park, E.; Daley, J.F.; Schlossman, S.F. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J. Immunol. 1983, 131, 244–250. [Google Scholar] [PubMed]
- De Oliveira, S.N.; Wang, J.; Ryan, C.; Morrison, S.L.; Kohn, D.B.; Hollis, R.P. A CD19/Fc fusion protein for detection of anti-CD19 chimeric antigen receptors. J. Transl. Med. 2013, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Roberts, Z.J.; Better, M.; Bot, A.; Roberts, M.R.; Ribas, A. Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk. Lymphoma 2018, 59, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharm. 2021, 139, 111605. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. 2021, 12, 81. [Google Scholar] [CrossRef]
- George, K.; Woollett, G. Insulins as drugs or biologics in the USA: What difference does it make and why does it matter? BioDrugs 2019, 33, 447–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kow, C.S.; Hasan, S.S. Optimal time for the resumption of biologics after COVID-19. JAAD Int. 2020, 1, 189. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [CrossRef]
- Su, Z.; Wang, B.; Almo, S.C.; Wu, Y. Understanding the targeting mechanisms of multi-specific biologics in immunotherapy with multiscale modeling. iScience 2020, 23, 101835. [Google Scholar] [CrossRef] [PubMed]
- Glaesner, W.; Vick, A.M.; Millican, R.; Ellis, B.; Tschang, S.H.; Tian, Y.; Bokvist, K.; Brenner, M.; Koester, A.; Porksen, N.; et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab. Res. Rev. 2010, 26, 287–296. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.R. Fusion-proteins as biopharmaceuticals—Applications and challenges. Curr. Opin. Drug Discov. Devel. 2009, 12, 284–295. [Google Scholar] [PubMed]
- Strohl, W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A. Combination approaches and anti-PD1 therapies: The focus of new research at ESMO and SMR. Melanoma Manag. 2015, 2, 9–14. [Google Scholar] [CrossRef]
- Greenblatt, K.; Khaddour, K. Trastuzumab; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hinner, M.J.; Aiba, R.S.B.; Jaquin, T.J.; Berger, S.; Durr, M.C.; Schlosser, C.; Allersdorfer, A.; Wiedenmann, A.; Matschiner, G.; Schuler, J.; et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin. Cancer Res. 2019, 25, 5878–5889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, C.; Su, L.; Wu, L.; Dufort, F.J.; Sanford, T.; Birt, A.; Hackel, B.J.; Hombach, A.; Abken, H.; Lobb, R.R.; et al. Anti-CD19 CAR T cells potently redirected to kill solid tumor cells. PLoS ONE 2021, 16, e0247701. [Google Scholar] [CrossRef] [PubMed]
- Martyniszyn, A.; Krahl, A.C.; Andre, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 Bispecific CAR T Cells for the treatment of B-cell malignancies. Hum. Gene 2017, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Ueda, T.; Kaneko, S. In Vitro Differentiation of T Cell: From CAR-Modified T-iPSC. Methods Mol. Biol. 2019, 2048, 85–91. [Google Scholar] [CrossRef]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kuhnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 5088. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Musial-Siwek, M.; Karabadzhak, A.; Andreev, O.A.; Reshetnyak, Y.K.; Engelman, D.M. Tuning the insertion properties of pHLIP. Biochim. Biophys. Acta 2010, 1798, 1041–1046. [Google Scholar] [CrossRef] [Green Version]
- Reshetnyak, Y.K.; Andreev, O.A.; Segala, M.; Markin, V.S.; Engelman, D.M. Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 15340–15345. [Google Scholar] [CrossRef] [Green Version]
- An, M.; Wijesinghe, D.; Andreev, O.A.; Reshetnyak, Y.K.; Engelman, D.M. pH-(low)-insertion-peptide (pHLIP) translocation of membrane impermeable phalloidin toxin inhibits cancer cell proliferation. Proc. Natl. Acad. Sci. USA 2010, 107, 20246–20250. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Luna, I.; Persad, K.L.; Agopsowicz, K.; Jay, D.A.; West, F.G.; Hitt, M.M.; Persad, S. Inhibition of triple negative breast cancer metastasis and invasiveness by novel drugs that target epithelial to mesenchymal transition. Sci Rep. 2021, 11, 11757. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Feng, K.; Tong, C.; Jia, H.; Liu, Y.; Wang, Y.; Ti, D.; Yang, Q.; Wu, Z.; Han, W. Efficiency and side effects of anti-CD38 CAR T cells in an adult patient with relapsed B-ALL after failure of bi-specific CD19/CD22 CAR T cell treatment. Cell Mol. Immunol. 2020, 17, 430–432. [Google Scholar] [CrossRef]
- Cordoba, S.; Onuoha, S.; Thomas, S.; Pignataro, D.S.; Hough, R.; Ghorashian, S.; Vora, A.; Bonney, D.; Veys, P.; Rao, K.; et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: A phase 1 trial. Nat. Med. 2021, 27, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Yang, X.; Yang, F.; Wang, T.; Ding, L.; Song, L.; Miao, Y.; Wang, X.; Ma, Y.; Luo, C.; et al. Outcomes of Anti-CD19 CAR-T treatment of pediatric B-all with bone marrow and extramedullary relapse. Cancer Res. Treat. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Guo, Y.; Wang, Y.; Wu, Z.; Bo, J.; Zhang, B.; Zhu, J.; Han, W. Clinical development of CAR T cell therapy in China: 2020 update. Cell Mol. Immunol. 2021, 18, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.D.; Slater, K.J.; Crouch, S.P.M. Changes in intracellular adp:Atp ratios as a marker of apoptosis. Sci. World J. 2001, 1, 58. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.K.; Kleinheinz, T.L.; Peterson, D.; Moffat, J.G. A simple high-content cell cycle assay reveals frequent discrepancies between cell number and ATP and MTS proliferation assays. PLoS ONE 2013, 8, e63583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, E.T.A.; Cimini, B.; Klemm, A.H.; Wahlby, C.; Carpenter, A.E.; Eliceiri, K.W. ImageJ and cell profiler: Complements in open-source bioimage analysis. Curr. Protoc. 2021, 1, e89. [Google Scholar] [CrossRef]
- Juanes, M.A.; Fees, C.P.; Hoeprich, G.J.; Jaiswal, R.; Goode, B.L. EB1 Directly regulates APC-mediated actin nucleation. Curr. Biol. 2020, 30, 4763.e4768–4772.e4768. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 2012, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Ke, N.; Berkmen, M. Use of the shuffle strains in production of proteins. Curr. Protoc. Protein Sci. 2016, 85, 5–26. [Google Scholar] [CrossRef]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- Lauko, A.; Mu, Z.; Gutmann, D.H.; Naik, U.P.; Lathia, J.D. Junctional Adhesion Molecules in Cancer: A Paradigm for the Diverse Functions of Cell-Cell Interactions in Tumor Progression. Cancer Res. 2020, 80, 4878–4885. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Brandl, A.; Mattenheimer, K.; Graf, C.; Ritz, M.; Ruckdeschel, A.; Stuhmer, T.; Mokhtari, Z.; Rudelius, M.; Dotterweich, J.; et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia 2018, 32, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Da Via, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores angiogenic homeostasis and suppresses tumor progression. Haematologica 2021, 106, 1943–1956. [Google Scholar] [CrossRef] [PubMed]
- Sachs, F. Mechanical Transduction and the Dark Energy of Biology. Biophys. J. 2018, 114, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, P.; Capaldo, C.T.; Koch, S.; Kolegraff, K.; Rankin, C.R.; Farkas, A.E.; Feasel, M.E.; Li, L.; Addis, C.; Parkos, C.A.; et al. JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep. 2011, 12, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, M.U.; Naik, T.U.; Suckow, A.T.; Duncan, M.K.; Naik, U.P. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008, 68, 2194–2203. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, R.; Selmi, A.; Wojkowska, D.; Karolczak, K.; Popielarski, M.; Stasiak, M.; Salifu, M.O.; Babinska, A.; Swiatkowska, M. Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res. Treat. 2020, 179, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magara, K.; Takasawa, A.; Osanai, M.; Ota, M.; Tagami, Y.; Ono, Y.; Takasawa, K.; Murata, M.; Hirohashi, Y.; Miyajima, M.; et al. Elevated expression of JAM-A promotes neoplastic properties of lung adenocarcinoma. Cancer Sci. 2017, 108, 2306–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Vleminckx, K.; Vakaet, L., Jr.; Mareel, M.; Fiers, W.; van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-cadherin inhibits CD103 antitumor activity and reduces checkpoint blockade responsiveness in melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ye, M.; Zhou, J.; Wang, Z.W.; Zhu, X. Restoring E-cadherin expression by natural compounds for anticancer therapies in genital and urinary cancers. Mol. Oncolytics 2019, 14, 130–138. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza, C.; Mizrachi, D. Using the Power of Junctional Adhesion Molecules Combined with the Target of CAR-T to Inhibit Cancer Proliferation, Metastasis and Eradicate Tumors. Biomedicines 2022, 10, 381. https://doi.org/10.3390/biomedicines10020381
Mendoza C, Mizrachi D. Using the Power of Junctional Adhesion Molecules Combined with the Target of CAR-T to Inhibit Cancer Proliferation, Metastasis and Eradicate Tumors. Biomedicines. 2022; 10(2):381. https://doi.org/10.3390/biomedicines10020381
Chicago/Turabian StyleMendoza, Christopher, and Dario Mizrachi. 2022. "Using the Power of Junctional Adhesion Molecules Combined with the Target of CAR-T to Inhibit Cancer Proliferation, Metastasis and Eradicate Tumors" Biomedicines 10, no. 2: 381. https://doi.org/10.3390/biomedicines10020381
APA StyleMendoza, C., & Mizrachi, D. (2022). Using the Power of Junctional Adhesion Molecules Combined with the Target of CAR-T to Inhibit Cancer Proliferation, Metastasis and Eradicate Tumors. Biomedicines, 10(2), 381. https://doi.org/10.3390/biomedicines10020381